




Abstract
Familial medullary thyroid cancer (MTC) and its precursor, C cell hyperplasia (CCH), is associated with germline RET mutations causing multiple endocrine neoplasia type 2. However, some rare families with apparent MTC/CCH predisposition do not have a detectable RET mutation. To identify novel MTC/CCH predisposition genes we undertook exome resequencing studies in a family with apparent predisposition to MTC/CCH and no identifiable RET mutation. We identified a novel ESR2 frameshift mutation, c.948delT, which segregated with histological diagnosis following thyroid surgery in family members and demonstrated loss of ESR2-encoded ERβ expression in the MTC tumour. ERα and ERβ form heterodimers binding DNA at specific oestrogen-responsive elements (EREs) to regulate gene transcription. ERβ represses ERα-mediated activation of the ERE and the RET promoter contains three EREs. In vitro, we showed that ESR2 c.948delT results in unopposed ERα mediated increased cellular proliferation, activation of the ERE and increased RET expression. In vivo, immunostaining of CCH and MTC using an anti-RET antibody demonstrated increased RET expression. Together these findings identify germline ESR2 mutation as a novel cause of familial MTC/CCH and provide important insights into a novel mechanism causing increased RET expression in tumourigenesis.
Introduction
Thyroid cancer is the most common endocrine malignancy. Although medullary thyroid cancer (MTC), arising from the calcitonin secreting para-follicular cells, only accounts for ∼5% of thyroid cancers (1), nearly 50% of patients present with Stage III or Stage IV disease and only 21% of patients presenting with Stage IV disease survive 10 years (2).
In ∼25% of MTC, there is a germline RET mutation (3) predisposing to multiple endocrine neoplasia type 2 (MEN2), a dominantly inherited endocrine tumour predisposition syndrome characterized by predisposition to the development of young onset MTC and often primary C cell hyperplasia (CCH), a precursor to MTC (4). Other features may also occur such as phaeochromocytoma, primary hyperparathyroidism (HPT) and, rarely, developmental abnormalities, for example, a marfanoid habitus and ganglioneuromatosis of the mouth and gut (4).
The majority of RET mutations predisposing to MEN2 result in single amino acid substitutions affecting key residues in the extra-cellular and kinase domains of the RET receptor (5, 6) causing inappropriate constitutive RET activation. Detection of a germline RET mutation, enables pre-symptomatic interventions such as prophylactic thyroidectomy to be offered to at-risk gene carriers (6). Thus all individuals presenting with MTC or primary CCH should be offered germline RET testing (7); however, some families with an apparent predisposition to MTC/CCH do not harbour a germline RET gene alteration (8, 9) suggesting that further predisposing gene alterations remain to be identified. We investigated a kindred with non-RET MTC/CCH and detected a novel constitutional frameshift mutation in ESR2 encoding the beta subunit of the oestrogen receptor, ERβ (10).
Results
Case report
The index case presented age 22 years with MTC (Supplementary Material, Fig. S1). Although no constitutional mutation of exons 10, 11 or 13–16 of RET was detected, in view of the young age of diagnosis his monozygotic twin brother was offered follow-up and underwent thyroidectomy age 33 years because of abnormal pentagastrin stimulation testing; histological review showed the presence of CCH. Prophylactic thyroid surgery was then undertaken in the offspring of both brothers (individuals III:1–5); individuals III:2 and III:3 were found to have CCH and individuals III:1, III:4 and III:5 to have normal thyroid tissue. Pentagastrin stimulation testing was subsequently offered to the wider family and individual I:1 noted to have mildly abnormal results. In the index case, further constitutional molecular genetic testing did not detect any mutations in the remaining exons of RET, karyotype analysis was normal and array CGH analysis did not demonstrate any clinically significant CNVs (data not shown). We therefore hypothesised a novel gene alteration was predisposing to MTC/CCH in the family and undertook exome sequencing in two affected members (II:3 and III:3).
Identification of germline ESR2 mutation in familial MTC/CCH
We filtered the variants identified using the dbSNP (11), NHLBI Exome Variant Server (EVS) (12) and 1000 genomes (13) datasets looking for potentially disrupting variants present in both samples, prioritizing frameshift and nonsense changes. We did not identify any deleterious alterations in the genes known to be mutated in hereditary cancer predisposition (14, Supplementary Material, Table S1) but did identify a novel frameshift alteration of ESR2 (c.948delT; p.Gly318Alafs*22) present in both affected individuals but not in the NHLBI EVS nor in 2577 individuals from the 1000 genome project (12, 13).
Constitutional ESR2 sequencing in other family members demonstrated the c.948delT variant to be present in the two family members found to have CCH (individuals II:1 and III:2) but not in the three (individuals III:1, III:4, III:5) with normal thyroid histology (Fig. 1A, Supplementary Material, Fig. S1).
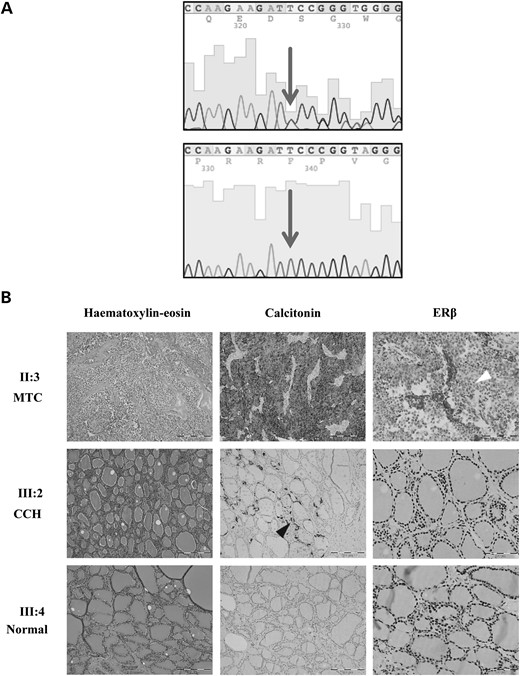
Germline ESR2 mutation detected in family members with MTC/CCH and loss of ERβ staining in associated MTC tumour. (A) Sanger sequencing traces showing the presence of ESR2 c.948delT (upper panel) and corresponding normal trace (lower panel). Affected nucleotide is indicated by an arrow. (B) Representative histological stains in sections following thyroid surgery in representative family members II:3 (MTC), III:2 (CCH), III:4 (normal thyroid). Haematoxylin and eosin stain showing MTC (upper panel) and apparently normal thyroid tissue (middle and lower panels). Calcitonin staining demonstrating MTC (upper panel), areas of CCH within apparently normal thyroid tissue (black arrowhead, middle panel) and normal thyroid tissue (lower panel). ERβ immunohistochemistry showing loss of nuclear expression in the MTC (upper panel, white arrowhead). Middle and lower panels—preserved nuclear staining of ERβ in follicular epithelial cells. It was not possible to determine if there was loss of ERβ staining associated with areas of CCH (middle panel).
Loss of ERβ staining in ESR2 c.948delT-associated MTC
To investigate the role of ESR2 in MTC tumourigenesis we initially undertook immunohistochemistry using an anti-ERβ antibody and demonstrated complete loss of nuclear staining in the c.948delT-associated MTC (Fig. 1B) with retained staining in the corresponding normal thyroid tissue. However, despite direct sequencing of the complete ESR2 coding region and loss of heterozygosity (LOH) studies using closely linked microsatellite markers in archived tumour material, we did not detect a somatic alteration of ESR2 (data not shown). Whole genome copy number analysis by molecular inversion probe technology did not reveal any pathological CNVs affecting either chromosomes 14 or 10 (harbouring the ESR2 and RET loci, respectively) (Supplementary Material, Fig. S2).
No additional constitutional ESR2 mutations in other cases of apparent MTC predisposition
To ascertain whether constitutional alterations of ESR2 might be involved in other cases of apparent MTC predisposition, we undertook sequencing of the coding region of ESR2 in 19 individuals with apparently isolated MTC, 8 of whom were diagnosed <40 years of age (mean age at diagnosis 47.5 years, range 33–71 years, SEM 2.96). The entire coding region of RET had been previously sequenced and no alterations detected other than known polymorphisms. We identified a novel germline missense alteration, c.382G>C; p.V128L, in a female who developed an apparently isolated MTC age 36 years (Fig. 2A). This variant was also detected in a pair of siblings, one of whom had a papillary thyroid cancer age 60 years and the other a MTC age 57 years. We did not detect any constitutional ESR2 alterations, other than known polymorphisms, in a further three families with familial non-RET MTC (data not shown).
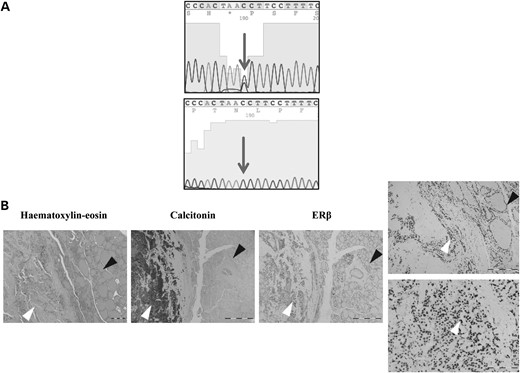
Germline ESR2 alteration detected in an individual with early onset MTC and preserved ERβ staining in associated MTC tumour. (A) Sanger sequencing traces showing constitutional ESR2 alteration c.382G>C (upper panel) and corresponding normal trace (lower panel) detected in an individual with apparently isolated MTC age 36 years. Affected nucleotide is indicated by an arrow. (B) Representative histological stains in sections following thyroid surgery. Left, haematoxylin and eosin stain showing MTC (white arrowhead) and corresponding normal thyroid tissue (black arrowhead). Middle, calcitonin stain demonstrating MTC (white arrowhead) with corresponding unstained normal thyroid tissue (black arrowhead). Right, ERβ immunohistochemistry showing preserved nuclear expression in both MTC (white arrowhead and high power insert) and corresponding normal thyroid tissue (black arrowhead and high power insert).
Further investigation using archived tumour material from the individual with the apparently isolated MTC and ESR2 c.382G>C (no blocks were available from the sibling pair) revealed preserved ERβ staining (Fig. 2B) and no somatic ESR2 sequence alterations, LOH or pathological CNVs of chromosomes 14 or 10 were detected. Although this variant was not present in the NHLBI EVS (12) or 1000 genome project (13) datasets, further analysis using SIFT (15) predicted it to be tolerated, and PolyPhen-2 (16) to be benign; we were therefore unable to exclude that ESR2 c.382G>C may represent a rare polymorphism and did not evaluate further.
No somatic mutations of ESR2 detected in sporadic MTC
As hereditary cancer predisposing genes are often implicated in sporadic tumours (17), we investigated whether somatic mutations of ESR2 also occurred in sporadic MTC. However, direct sequencing of ESR2 in 15 fresh frozen sporadic MTC did not reveal any alterations other than known polymorphisms (data not shown). Whilst the cosmic (18) and TCGA data sets (19), containing data for over 21 000 tumours, did not have any data regarding ESR2 in MTC, we did note the presence of somatic ESR2 mutations in other tumours of neuroectodermal origin, for example glioma and melanoma.
ESR2 c.948delT is associated with unrestrained ERα-driven cell proliferation
We then sought to understand how this rare novel ESR2 frameshift alteration might cause tumourigenesis. Initially we determined stability of the ESR2 c.948delT mutant compared with wild-type (wt) ESR2. Transient transfection of HCT116 cells, followed by the addition of the transcription inhibitor, actinomycin, resulted in significantly lower ESR2 c.948delT mRNA levels over a 24 h time period compared with wtESR2 indicating reduced stability of the mutant mRNA (Supplementary Material, Fig. S3).
ESR2-encoded ERβ forms either homo- or, preferentially, hetero-dimers with ESR1-encoded ERα to bind DNA at specific oestrogen-responsive elements (EREs) within the promoters of target genes to regulate transcription (20). ERα is the more potent activator and ERβ can repress ERα; thus transcriptional activity is determined by the relative ERα/ERβ proportion (21–23).
We next investigated whether ESR2 c.948delT altered cellular growth. In MCF-7 cells, ESR2 c.948delT failed to restrain the ERα-driven proliferation of MCF-7 cells in response to either 17β-estradiol (E2) or the ERα selective agonist, PPT. In contrast, wtESR2 was capable of inhibiting cell proliferation by up to 30% in both E2 and PPT treated MCF-7 cells over-expressing ERα (Fig. 3).
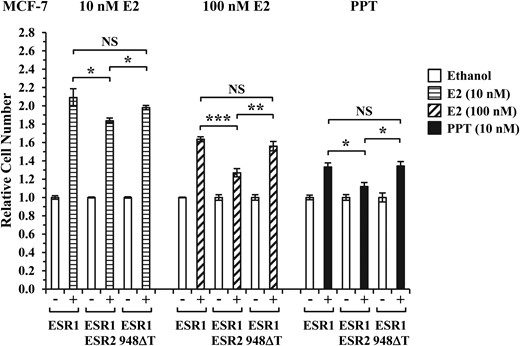
ESR2 c.948delT is associated with unrestrained ERα-driven cellular proliferation. MCF-7 cells were reverse transfected with wtESR1 (ESR1) alone or in combination with either wtESR2 (ESR2) or ESR2 c.948delT (948ΔT) for 24 h and then treated with 10 nM E2, 100 nM E2 or 10 nM PPT for 72 h, compared with vehicle (ethanol) treated controls. Cell proliferation status was quantified by luciferase assay using the CellTiter-Glo Luminescent Cell Viability Assay (Promega) and expressed relative to ethanol treated controls. Data presented as mean values ± SE (n = 4). *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant.
Loss of ERβ function causes increased ERE activity and RET expression
In both HCT116 and MCF-7 cells, in response to either E2 or PPT agonist, wtESR1 activated the ERE, as indicated by increased luciferase expression, and this was significantly restrained in the presence of wtESR2. However, co-transfection of wtESR1 with ESR2 c.948delT restored expression to a level similar to that seen with wtESR1 alone indicating that the ESR2 c.948delT mutant has lost the ability to restrain ERα-mediated activation of the ERE (Fig. 4).
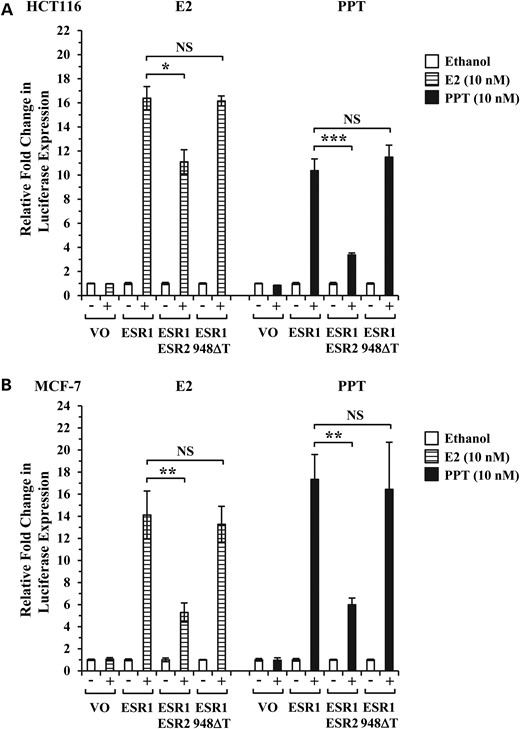
Diminished ability of ESR2 c.948delT to inhibit wtESR1. Luciferase assays evaluating transcriptional activity of a transfected ERE by vector only (VO), wtESR1 (ESR1) alone or in combination with either wtESR2 (ESR2) or ESR2 c.948delT (948ΔT) mutant at a ratio of 1:3 in (A) HCT116 and (B) MCF-7 cell lines. Cells were treated with 10 nM E2, 10 nM PPT or ethanol control as indicated for 24 h prior to cell lysis and luciferase activity measurement. Relative fold change presents mean values ± SE from at least two independent experiments (n = 4 wells per experiment). *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant.
We then investigated how inappropriate activation of the ERE by c.948delT might cause MTC tumourigenesis. Inappropriate up-regulation of RET activity causing MTC is well established (4) and we noted that the RET promoter (−34 to −314) contains three ERE (24). Furthermore, in in vitro studies, RET has been established as an ER target gene (25–27). We therefore investigated whether loss of ERβ function might lead to up-regulation of RET expression.
As MCF-7 cells have intrinsic ER activity, we used HCT116 cells to investigate the effect of ESR2 c.948delT on RET expression. Co-transfection of wtESR1 with wtESR2 in HCT116 cells, in the presence of either E2 or PPT, resulted in decreased RET expression when compared with wtESR1 alone; however, over-expression of wtESR1 and ESR2 c.948delT, restored RET expression to a level similar to that seen with wtESR1 alone (Fig. 5A and B).
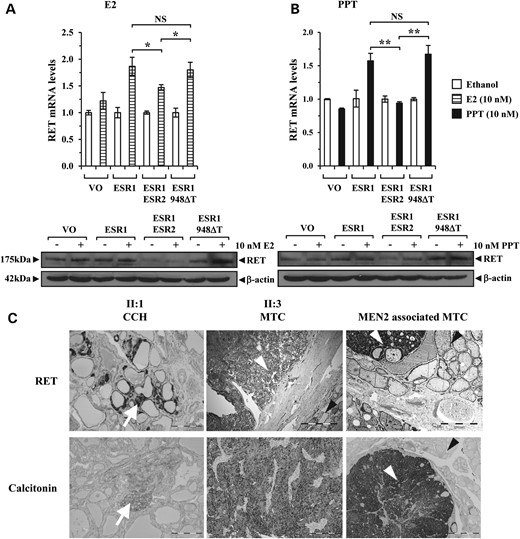
ESR2 c.948delT is associated with elevated RET expression. (A and B) real-time PCR (upper panel) and western blot (lower panel) analyses of RET expression levels in HCT116 cells transfected with wtESR1 (ESR1) alone or in combination with either wtESR2 (ESR2) or ESR2 c.948delT (948ΔT) and then treated with either (A) 10 nM E2 (+) or (B) 10 nM PPT (+) compared with ethanol control (−). Real-time data presented as mean RET mRNA levels ± SE (n = 4). In control experiment cells were transfected with vector only (VO) as indicated. (C) Representative histological sections showing immunohistochemical staining for RET (upper panels) with corresponding calcitonin stains (lower panels) to confirm the presence of CCH/MTC. Left—archived slides from individual II:1 showing areas of intense RET staining (white arrow, upper panel) and area of marked calcitonin staining indicating the presence of CCH (white arrowhead, lower panel) within normal thyroid tissue. Middle—MTC tumour from individual II:3 showing intense RET expression (white arrowhead, upper panel) when compared with corresponding normal thyroid tissue (black arrowhead, upper panel). Calcitonin stain (lower panel) confirms the presence of MTC (same panel as in Fig. 1B). Right—MEN2-associated MTC (white arrowhead, upper panel) also showing intense RET expression when compared with corresponding normal thyroid tissue (black arrowhead, upper panel), for comparison with middle panel. Calcitonin stain (lower panel) confirms the presence of MTC (white arrowhead, lower panel) with corresponding normal thyroid tissue also shown (black arrowhead, lower panel).
In vivo, RET immunostaining using archived sections from the ESR2 c.948delT-associated MTC (individual II:3) and analysis of archived stained slides showing CCH (individual II:1) demonstrated increased RET staining (Fig. 5C). In particular, the intensity of RET expression in the MTC was stronger than in corresponding normal thyroid tissue and similar to that in an MEN2-associated MTC with a known constitutional RET mutation (Fig. 5C). Analysis of RET exons 10, 11 and 13–16 in the ESR2 c.948delT-associated MTC did not detect a somatic mutation to account for the increased expression (data not shown).
Discussion
By targeted capture array-based exome sequencing we detected a novel loss of function ESR2 mutation in a family with predisposition to MTC/CCH. As the interpretation of novel rare variants identified through next generation sequencing approaches in isolated families is challenging, we sought functional evidence to establish pathogenicity.
Initially, we demonstrated tumour specific loss of ERβ expression in the c.948delT-associated MTC which we considered indicative of loss of function as ERβ is known to be expressed by human para-follicular C cells (28). Whilst ESR2 knockout mice display a phenotype more in keeping with a reduced fertility phenotype in females rather than one associated with tumourigenesis (29), ESR2 has been shown to inhibit cell proliferation in vitro (30) and tumour formation in nude mice (31). Although we did not identify a somatic ESR2 alteration in this tumour and were unable to investigate ESR2 promoter hypermethylation, we note that ESR2 promoter hypermethylation associated with ERβ down-regulation has been detected in a variety of tumour types (32–34). As with other cancer predisposition syndromes (35), it is also probable that additional somatic mutations are required in the transition from normal thyroid to MTC and we did detect low level loss of chromosomes 3 and 13.
It is likely that ERβ loss of function in MTC is rare as we did not detect loss of ERβ expression in 12 apparently sporadic MTC tumours nor an intragenic ESR2 mutation in 15 sporadic MTCs. Interestingly, studies of MTC have indicated that RAS and RET are the dominant driver pathways in tumourigenesis, with few mutations being detected in other genes and none in ESR2 (36). Our findings are also in keeping with large scale studies of sporadic tumours where no CNVs, and only one putative mutation of ESR2 (missense alteration in a single papillary thyroid cancer), have been detected in thyroid tumours (18, 19). Such relative infrequency of somatic mutation in sporadic tumours in genes predisposing to hereditary cancers is well recognized, for example the low rate of somatic BRCA1 and BRCA2 mutations in sporadic breast cancer (37, 38). We did note that somatic ESR2 mutations have been detected in other neuroectodermal derived tumours such as glioma and melanoma and also breast cancer samples indicating that inappropriate up-regulated ERα activity may also be involved in their tumourigenesis (18, 19).
We did not detect a constitutional ESR2 mutation in a further three families with an apparent genetic predisposition to MTC and CCH and it is likely that, akin to other familial cancer predisposition syndromes (39), there is also heterogeneity in the genetic predisposition to MTC/CCH.
The association of increased RET activity with MTC tumourigenesis is well established (4) and here we demonstrate a novel mechanism of MTC tumourigenesis whereby loss of ERβ function results in ERα-driven RET expression, likely mediated through the ERE on the RET promoter. Whilst there may be an unidentified constitutional RET mutation, or another mechanism of RET up-regulation, we have also shown ESR2 c.948delT to cause increased cellular proliferation indicating a likely role in tumourigenesis; however, we cannot exclude that other mechanisms of ERβ action may also be involved, for example through loss of ERβ mediated inhibition of HIF-1α causing inappropriate HIF pathway activation (40).
Although we detected a mutation in just one family, our results suggest that constitutional ESR2 mutation is of high penetrance for CCH/MTC. However, as individual I:1 has not overtly presented with MTC, we cannot exclude that there may be variable penetrance, as occurs for germline RET mutations and in other familial cancer predisposition syndromes such as paraganglioma-phaeochromocytoma predisposition (4, 41).
It is interesting to speculate whether constitutional ESR2 mutations are also associated with predisposition to HPT and phaeochromocytoma. These tumours have not occurred to date in the family (relevant clinical and laboratory investigation has been undertaken) and whole exome sequencing of both constitutional DNA from individuals with apparent genetic predisposition to phaeochromocytoma (12 individuals) and sporadic phaeochromocytoma (7 tumours) has not detected any deleterious ESR2 mutations (ERM unpublished observations). Nonetheless, given the known tumour predisposition associated with dysregulated RET activity, it would seem prudent to offer clinical surveillance for phaeochromocytoma and HPT, and prophylactic thyroidectomy, where a loss of function constitutional ESR2 mutation is detected.
The investigation of rare familial cancer predisposition often provides important insights into cell biology with the potential for novel therapeutic approaches (42) and our findings also provide further evidence of the interaction between ERα and RET in tumourigenesis. Many invasive breast cancers expressing ERα also express RET (4) and in recent pre-clinical models of breast cancer, targeting both RET and ERα decreases the growth and metastatic potential of breast cancer cells (43). Thus tumours associated with loss of ERβ function might be suitable candidates for therapeutic agents targeting the RET pathway (44), RET tyrosine kinase active site (45) or ERα (46).
In summary, our data are the first to identify a novel genetic cause for familial MTC since the identification of the RET gene. We demonstrate a novel mechanism promoting up-regulation of RET activity through constitutional ESR2 mutation which provides opportunities for pre-symptomatic testing and potential novel therapeutic strategies.
Materials and Methods
Whole exome sequencing analysis
Exome sequencing was performed at the Biomedical Research Centre at Kings College London as previously described (47).
Sanger sequencing
Genomic DNA was extracted from formalin-fixed paraffin embedded (FFPE) tissue using the QIAamp DNA FFPE kit (Qiagen) and from peripheral leucocytes using the Nucleon BACC2 kit (Amersham Biosciences) (sequences and PCR conditions available upon request).
Immunohistochemistry
Slides were de-paraffinized using Benchmark Special Stains Deparaffinization Solution. Heat-induced antigen retrieval was performed using EDTA buffer as previously described (48). Immunostaining was performed using the Ventana Benchmark Automated Slide Stainer (Ventana Medical Systems) with antibodies against ERβ (Leica Biosystems, 1:50), calcitonin (Dako IR515) and RET (Abcam EPR2871, 1:500).
LOH analyses
Microsatellite markers were used to determine LOH at the ESR2 locus on chromosome 14q23.2: one, previously described (49) within intron six (chr14:64 720 279–64 720 323) and one 5′ upstream (chr14:64 802 692–64 802 742) (sequences available upon request). PCR amplification was performed on genomic DNA templates using fluorescently labelled primers. PCR products were electrophoresed on an ABI Genetic Analyzer and analysed by GeneMapper fragment analysis software (Applied Biosystems).
Oncoscan® FFPE assay kit
The OncoScan® FFPE assay kit platform was utilized to identify whole genome copy number variations (CNV) as previously described (50).
Cell culture
In the absence of a suitable MTC cell line we used HCT116 (Health Protection Agency Culture Collections, UK) and MCF-7 cells (European Collection of Animal Cell Cultures) cultured in McCoy′s 5A (Life Technologies) with 5% fetal bovine serum (Invitrogen), penicillin (104 U/ml) and streptomycin (104 mcg/ml) (Invitrogen) for HCT116 cells and RPMI 1640 (Life Technologies) with 5% fetal bovine serum (Invitrogen), penicillin (105 U/l) and streptomycin (100 mg/l) for MCF-7 cells. Cells were treated with 17β-oestradiol (E2, Sigma) and 4,4′,4″-(4-propyl-[1H]-pyrazole-1,3,5-triyl(trisphenol) (PPT, Tocris) at final concentrations of 10 and 100 nM, in phenol red-free medium (Life Technologies) with 5% charcoal-stripped serum.
Transfection
The ESR1/2 ORFS were cloned into Sgf1 and Mlu1 restriction sites of pCMV6-AN-Myc (Origene). QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) was used to delete the thymine at position 948 of ESR2, generating ESR2, c.948delT. Plasmid DNA transfections were performed with TransIT LT1 (Mirus Bio LLC). In some experiments, plasmid DNA was introduced into cells by reverse transfection (lipid: DNA complexes plated prior to addition of cells).
RNA extraction, real-time PCR and mRNA stability assay
Total RNA was extracted and reverse transcribed using RNeasy Micro Kit (Qiagen) and Reverse Transcription System (Promega) with expression determined by the 7500 Real-time PCR system (Applied Biosystems). qRT-PCR was carried out on 96-well plates using FAM-labelled Taqman probes (Life Technologies) for ESR2 (Hs01100359_m1), RET (Hs01120030_m1) and PPIA (Hs04194521_s1). In mRNA stability experiments, HCT116 cells were treated with actinomycin D at a final concentration of 10 µg/ml for 4, 8 and 24 h prior to total RNA extraction.
Western blotting
Western blots, performed as described previously (51), were probed with antibodies against RET (C31B4, Cell Signaling Technology), 1:1000; ERβ (H150, Santa Cruz Biotechnology), 1:1000; c-Myc, 1:1000 and β actin; 1:10 000 (Sigma-Aldrich). Antigen–antibody complexes were detected using ECL Plus (Amersham Biosciences).
Luciferase reporter assays
Luciferase reporter assays were performed using the Cignal ERE Reporter Kit (Qiagen). HCT116 (7500 cells/well) and MCF-7 (15 000 cells/well) cells were reverse transfected with 50 ng Cignal ERE reporter along with 100 ng ER expression vectors with TransIT LT1. After 24 h hormone incubation, cells were harvested with lysis buffer (Promega) and the firefly and renilla luciferase activities determined with a dual luciferase assay kit (Promega), measuring luminescence with a Centro LB960 microplate luminometer (Berthold Technologies).
Cell proliferation
Proliferation of MCF-7 cells was determined using the Celltiter-Glo® Luminescent Cell Viability Assay (Promega). In brief, 12 500 MCF-7 cells were reverse transfected with ER expression vectors and TransIT LT1 in 96 wells and then treated with hormones as indicated for 72 h prior to analysis of the luminescent signal using the Centro LB960 microplate luminometer (Berthold Technologies).
Statistical analyses
Data are displayed as mean ± SE. Normally distributed data were analysed using a two-tailed Student′s t test, unless otherwise indicated. A P < 0.05 was considered to be statistically significant.
Funding
The study was funded by the Queen Elizabeth Hospital Birmingham Charity and The Get A-Head Charitable Trust with support from Affymetrix UK Limited, the Technology Strategy Board (now Innovate-UK) Stratified Medicine Innovation Platform (#101032), the Canadian Institutes for Health Research (#142303), the Terry Fox Research Institute Transdisciplinary Training Program in Cancer Research and the National Institute for Health Research.
Acknowledgements
This manuscript is dedicated to the memory of Dr. Louise Brueton who undertook all the initial clinical work-up in the family. We are grateful to the families and patients for participation in the study.
Conflict of Interest statement. None declared.
References
Author notes
†Joint first authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}