




Abstract
Mutations of MYH9 , the gene for non-muscle myosin heavy chain IIA (NMMHC-IIA), cause a complex clinical phenotype characterized by macrothrombocytopenia and granulocyte inclusion bodies, often associated with deafness, cataracts and/or glomerulonephritis. The pathogenetic mechanisms of these defects are either completely unknown or controversial. In particular, it is a matter of debate whether haploinsufficiency or a dominant-negative effect of mutant allele is responsible for hematological abnormalities. We investigated 11 patients from six pedigrees with different MYH9 mutations. We evaluated NMMHC-IIA levels in platelets and granulocytes isolated from peripheral blood and in megakaryocytes (Mks) cultured from circulating progenitors. NMMHC-IIA distribution in Mks and granulocytes was also assessed. We demonstrated that all the investigated patients had a 50% reduction of NMMHC-IIA expression in platelets and that a similar defect was present also in Mks. In subjects with R1933X and E1945X mutations, the whole NMMHC-IIA of platelets and Mks was wild-type. No NMMHC-IIA inclusions were observed at any time of Mk maturation. In granulocytes, the extent of NMMHC-IIA reduction in patients with respect to control cells was significantly greater than that measured in platelets and Mks, and we found that wild-type protein was sequestered within most of the NMMHC-IIA inclusions. Altogether these results indicate that haploinsufficiency of NMMHC-IIA in megakaryocytic lineage is the mechanism of macrothrombocytopenia consequent to MYH9 mutations, whereas in granulocytes a dominant-negative effect of mutant allele is involved in the formation of inclusion bodies. The finding that the same mutations act through different mechanisms in different cells is surprising and requires further investigation.
INTRODUCTION
The term MYH9 -related disease ( MYH9 -RD) defines a spectrum of autosomal dominant thrombocytopenias with giant platelets: May–Hegglin anomaly (MHA), Sebastian (SBS), Fechtner (FTNS) and Epstein syndrome (EPTS), caused by mutations in the MYH9 gene, encoding for the heavy chain of non-muscle myosin IIA (NMMHC-IIA) ( 1 – 9 ). Affected patients present, since birth, macrothrombocytopenia and cytoplasmic aggregates of NMMHC-IIA in granulocytes recognizable by specific antibodies (Abs) ( 5 , 9 , 10 ). These aggregates are in most cases also evident on May–Grünwald–Giemsa (MGG)-stained blood films as characteristic ‘Döhle-like’ inclusions. Moreover, patients present the risk of developing in childhood or adult life the additional clinical features of sensorineural hearing loss, cataracts and/or a glomerulonephritis that can lead to end-stage renal failure ( 6 , 9 , 11 ). A recent revaluation of a large case series failed to identify correlations between specific- MYH9 mutations and the clinical phenotypes in 40 patients from 19 unrelated families, thus suggesting that MHA, SBS, FTNS and EPTS are not allelic forms, but rather represent a unique disease with clinical symptoms variably expressed ( 9 ).
Non-muscle myosin IIA is ubiquitously expressed in cells and tissues ( 12 ), where it is thought to participate in many cell functions, including cytokinesis, cell motility and maintenance of cell shape ( 13 – 17 ). As other conventional myosins, this is a hexameric enzyme composed of two heavy chains and two pairs of light chains with regulatory function. Dimerization of the heavy chains gives rise to a polar structure. The two N-terminal portions form two globular heads responsible for ATPase and actin-binding activity (motor domains), and a neck that participates in light chain binding and amplification of mechanical forces. The C-terminal portions wrap each other to form a rod-like, coiled-coil tail that allows the myosin molecule to polymerize into bipolar filaments ( 14 , 18 ).
To date, 23 different MYH9 mutations have been identified in MYH9 -RD pedigrees ( 4 – 7 , 9 , 11 ). Most of them are missense mutations or small in frame deletions affecting either the motor head or the rod-tail domain, whereas nonsense or frameshift alterations are all located in the last coding exon. The mechanisms that lead from MYH9 mutations to the clinical picture of MYH9 -RD are still poorly defined at both molecular and cellular levels. Whereas macrothrombocytopenia is likely to derive from a defect of the mechanism that originates platelets from megakaryocytes (Mks) ( 1 , 19 – 22 ), the pathophysiology of hearing loss, cataracts and nephritis is completely unknown, even if abnormalities of glomerular podocytes and tubular epithelia, with clumped distribution of NMMHC-IIA have been observed in a single patient with end-stage renal failure ( 23 ).
Recently, different in vivo and in vitro experimental approaches have led to contrasting conclusions about the molecular mechanism causing the MYH9 -RD phenotype. In fact, some findings indicated that the mutant NMMHC-IIA interacts with the wild-type (WT) protein and exerts a dominant-negative effect ( 10 , 24 ). Another group suggested that the D1424N mutation of MYH9 could result in haploinsufficiency ( 25 ). In this study, we further investigated the consequences of MYH9 mutations on NMMHC-IIA behavior in hematopoietic cells of 11 patients from six MYH9 -RD families, each carrying a different mutation. To this end, we cultured Mks from peripheral blood (PB) progenitors and, whenever possible, measured their level of NMMHC-IIA, together with the NMMHC-IIA content of platelets and granulocytes. Moreover, we assessed the distribution of NMMHC-IIA within Mks and granulocytes. We concluded that MYH9 mutations act through different mechanisms in different hematopoietic lineages: haploinsufficiency in Mks is the common result of different MYH9 mutations and it is responsible for defective platelet formation, while in granulocytes a dominant-negative effect of mutant NMMHC-IIA is involved in the formation of the inclusion bodies.
RESULTS
Analysis of NMMHC-IIA levels in MYH9-RD platelets
Identical amounts of proteins from the whole platelet lysates (20 µg) were subjected to immunoblotting analysis with Abs to NMMHC-IIA (NMF6 Ab, which recognizes the head of the molecule), β-actin and α-tubulin. Representative images of immunoblots are reported in Figure 1 . As expected, similar amounts of β-actin and α-tubulin were observed in patients and controls (Fig. 1 and data not shown). However, the content of NMMHC-IIA resulted evidently reduced with respect to controls in all the 11 studied patients. For each subject, the levels of NMMHC-IIA were evaluated by densitometric analysis, and normalized upon the levels of β-actin, to calculate the NMMHC-IIA/β-actin ratio (N/A ratio). As reported in Table 1 , in MYH9 -RD subjects the N/A ratio resulted approximately one-half with respect to controls (median, 50%; range, 36–68%). Patients from families A and B, which carry the R1933X and E1945X mutations resulting in a premature truncation of NMMHC-IIA, were also investigated using the PR-B440P Ab that specifically recognizes an unique epitope (residues 1948–1960) of the deleted region ( 26 , 27 ). Thus, in these cases, PR-B440P Ab is able to recognize only the WT NMMHC-IIA ( 10 ). As shown in Table 1 , similar results were obtained with NMF6 and PR-B440P Abs in platelets from families A and B, suggesting that in these patients most or the whole detectable platelet NMMHC-IIA was a WT protein.
Outcome of in vitro cultures of Mks
The Mk recovery at the end of in vitro cultures from PB progenitors was assessed in all 11 MYH9 -RD patients and in 10 controls studied simultaneously. Mks were identified on the basis of the expression of lineage-specific marker CD61 plus cell morphology. Immunomorphological evaluation revealed that the percentage of CD61+ cells with Mk morphology on total cultured cells at day 12 was 5.05% (median, range 2.81–5.27) in MYH9 -RD cases and 4.81% (median, range 2.81–5.72) in controls. Mk recruitment at day 12 for each 1×10 6 plated PB mononuclear cells was 27.4×10 3 (median, range 18.5–41.9) in MYH9 -RD subjects and 24.4×10 3 (median, range 12.1–42.6) in controls ( P =0.55). Upon classifying Mks at day 12 according to standard criteria ( 28 ), no significant differences in the profile of morphological maturation were observed between patients and controls (data not shown).
Analysis of NMMHC-IIA levels and distribution in MYH9-RD Mks
To investigate the NMMHC-IIA content, cultured Mks were purified by immunomagnetic bead technique on the basis of the expression of lineage-specific antigen CD41. Results of cell separation were evaluated by microscopy examination after immunostaining for CD61. We obtained Mk specimens suitable for immunoblotting analysis in seven out of 11 patients (families A, B and F) and in four controls. In these cases, the frequency of CD61+ cells with Mk morphology ranged from 87 to 92% in patients (median, 89%) and from 86 to 95% in controls (median, 91%). According to standard morphological criteria ( 28 ), the MYH9 -RD Mks were classifiable as follows: stage I Mks 21.4±1.9%, stage II 24.9±1.2%, stage III 27.4±1.0% and stage IV 26.3±1.8% (means±SD). No differences in the profile of morphological maturation were found between cases and controls (data not shown). Immunoblotting analysis of Mk lysates with NMF6 Ab showed a reduced expression of NMMHC-IIA in all the examined patients with respect to controls. Representative examples of immunoblots are shown in Figure 2 . The calculated N/A ratio in MYH9 -RD subjects was about one-half of control conditions (median, 45%; range, 42–54%) (Table 1 ), consistent with the results obtained in platelets. In patients from families A and B, we obtained similar results with NMF6 and PR-B440P Abs in each Mk sample, thus indicating that all the NMMHC-IIA measurable in Mks was a WT form of the protein (Fig. 2 and Table 1 ).
We then investigated whether an aberrant aggregation of mutant NMMHC-IIA, similar to that observable in granulocytes, was present in any phase of in vitro maturation of MYH9 -RD Mks. Preliminary observations indicated that, in our in vitro system, the earliest Mks precursors were observable at day 5. Thus, in six patients with different mutations (A/2, B/2, C/1, D/2, E/1, F/2), we harvested cell samples at day 5, 7, 9 and 12, and double-stained them with an anti-CD61 and NMF6 Ab to study NMMHC-IIA distribution by confocal microscopy. The protein was expressed in all the CD61+ cells, including the earliest CD61+ precursors observable at day 5. The distribution of NMMHC-IIA was identical in MYH9 -RD and control specimens in all the CD61+ cells at all the examined days of culture, without any evidence for aggregates formation in the cytoplasm. Figure 3 shows a representative example of a stage I Mk at day 5 and a stage IV Mk at day 12 from patient A/2.
Analysis of NMMHC-IIA levels and distribution in MYH9-RD granulocytes
Granulocyte lysates were obtained in seven patients belonging to the families A, B, C, E and in four controls. Microscopy analysis of cell samples prior to lysis indicated that the polymorphonuclear granulocytes were 95–98% of total nucleated cells, with no significant differences between patients and controls. In each case, platelet contamination was not above the 5% of total cells. In all patients, immunoblotting analysis with the NMF6 Ab showed reduced levels of NMMHC-IIA compared to control samples (Fig. 4 ). The calculated N/A ratio for MYH9 -RD granulocytes was 32% of control conditions (median, range 18–54%) (Table 1 ). Interestingly, in MYH9 -RD granulocytes, the percentage of reduction of the N/A ratio with respect to control cells resulted significantly greater than that one measured in platelets ( P <0.01) and Mks ( P <0.01) of the same seven patients (two-tailed Student's t -test for paired values). As in platelets and Mks, also in granulocytes, the PR-B440P Ab detected the same levels of NMMHC-IIA as NMF6 Ab in patients expressing the truncated R1933X and E1945X proteins (Fig. 4 and Table 1 ).
In addition to standard immunocytochemistry to reveal NMMHC-IIA aggregates in granulocytes, in patients with R1933X and E1945X, we performed a double-labeling on PB smears with the PR-B440P and the NMF6 Abs. Microscopy analysis revealed that most of the NMMHC-IIA aggregates identified in granulocytes by the NMF6 Ab were also stained by the PR-B440P (Fig. 5 ). Only some aggregates with small size (diameter 0.8–2 µm) were recognized exclusively by the NMF6 Ab. These results demonstrate that, in these patients, WT NMMHC-IIA was sequestered in most of the granulocyte inclusion bodies.
DISCUSSION
Although several aspects of the human disorder deriving from MYH9 mutations are being revealed, the pathogenetic mechanisms responsible for its complex phenotype are still poorly defined. In particular, the effect of mutations on the behavior of non-muscle myosin IIA in cells is under debate because of conflicting results from previous investigations ( 10 , 24 , 25 ). Deutsch et al . ( 25 ) observed that platelets from patients with the D1424N mutation expressed 50% of the amount of NMMHC-IIA with respect to healthy subjects, whereas the MYH9 mRNA was stable. On this basis, they concluded that mutant NMMHC-IIA was degraded early after transduction and that the D1424N mutation could cause the MYH9 -RD phenotype through haploinsufficiency of MYH9 . However, the level of NMMHC-IIA in Mks was not investigated and therefore, it cannot be excluded that the reduced platelet content in NMMHC-IIA is the consequence of a defective transmission of the mutant protein from Mks to platelets. More recently, Franke et al . ( 24 ) investigated the in vitro aggregation of D1424N and three other mutations in the rod-tail domain of NMMHC-IIA, R1165C, R1933X and E1841K. They showed that the mutant tail fragments interfered with the proper assembly of WT paracrystal structures, thus indicating that these mutations exert a dominant-negative biochemical effect. This in vitro result does not necessarily imply that the same mechanism is operative in patients, as early in vivo degradation of mutant protein could prevent its interaction with the WT NMMHC-IIA and the resulting dominant-negative effect. However, the in vitro findings by Franke et al . are consistent with previous in vivo observations by Kunishima et al. , who showed that WT NMMHC-IIA localized within the leukocyte inclusions in patients with 5779delC, 5818delG or R1933X ( 10 ). Thus, evidences supporting either haploinsufficiency or dominant-negative effect of mutant allele have been obtained in different patients with different MYH9 mutations.
In the present study, we evaluated the NMMHC-IIA expression levels and distribution in hematopoietic cells from 11 patients belonging to six MYH9 -RD pedigrees with different MYH9 mutations: four missense amino acid substitutions (N93K in the head domain and R1165C, D1424 N and E1841K in the rod-tail domain) and two premature protein truncations (R1933X and E1945X).
We first investigated, by immunoblotting analysis, the levels of NMMHC-IIA in MYH9 -RD platelets. In all the investigated patients, we found a reduced expression of the protein, and the median NMMHC-IIA level measured in patients resulted 50% of healthy subjects. These findings demonstrate that a defective NMMHC-IIA expression in platelets is the common result of different MYH9 mutations, affecting either the motor head or the rod-tail domain, and suggest that it represents a distinctive feature of the disease. Moreover, in patients with R1933X or E1945X mutation, we detected similar levels of NMMHC-IIA using Abs against the head of the molecule (present in mutant and WT proteins) or the COOH-terminus of the protein (absent in mutant proteins), indicating that the mutant form was not significantly expressed.
We then analyzed for the first time the NMMHC-IIA content of MYH9 -RD Mks. In seven patients carrying the R1933X, E1945X or E1841K mutations, we obtained from PB progenitors the amount of purified Mks required for immunoblotting analysis. In all the analyzed patients, we measured about one-half NMMHC-IIA compared to healthy subjects, demonstrating that the reduced amount of platelet NMMHC-IIA derived from a similar defect in Mks. As in platelets also in Mks from patients with the R1933X or E1945X mutation immunoblotting analysis indicated that the whole detectable NMMHC-IIA was the WT form of the protein. Altogether, these results strongly favor the hypothesis that a mechanism of haploinsufficiency within megakaryocytic lineage is primarily responsible for the macrothrombocytopenia of MYH9 -RD.
The pathophysiology of thrombocytopenia of this condition has not yet been established definitively. Some evidences indicate that it derives from a defect of platelet production, because previous investigations demonstrated that platelet in vivo survival was normal, and splenectomy had not effect on platelet count ( 19 , 21 , 29 ). In this study, we observed that both the recruitment and the profile of morphological maturation of Mks cultured from PB progenitors of 11 MYH9 -RD patients were indistinguishable from that of healthy subjects. This finding is consistent with one previous case report describing a normal number and morphology of Mks in bone marrow smears of a FTNS patient ( 22 ). Our observation confirms that the NMMHC-IIA deficiency in Mks does not significantly interfere with their intrinsic capability to differentiate in mature forms, and therefore it indirectly suggests that the NMMHC-IIA defect is critical only for the final step of platelet formation. Further studies on MYH9 -RD megakaryocytopoiesis are required to confirm this hypothesis and to understand how the reduced content of NMMHC-IIA impairs platelet formation and release.
We then investigated, by immunoblotting analysis, the NMMHC-IIA levels in granulocytes from seven patients with four different MYH9 mutations. The median NMMHC-IIA level in MYH9-RD granulocytes was 32% of controls, and it resulted significantly lower than those obtained in both Mks and platelets (45 and 50% of controls, respectively). Moreover, like in Mks and platelets, only the WT protein was detectable in granulocytes from patients with the nonsense R1933X and E1945X mutations. A possible explanation for these findings originates from the results of analysis of NMMHC-IIA distribution. In cultured CD61+ cells at any phase of differentiation or maturation, NMMHC-IIA always had a homogeneous cytoplasmic distribution indistinguishable from that of controls. At the opposite, the NMMHC-IIA aggregates typical of MYH9 -RD were always observed in all patients' granulocytes. Moreover, most of these aggregates were recognized by the Ab against the COOH-terminus of NMMHC-IIA in subjects with the R1933X or R1945X mutations, indicating that the WT protein was present in inclusions, consistent with previous observations on patients with 5779delC, 5818delG or R1933X ( 10 ). Although no direct evidence of localization of mutant NMMHC-IIA in such aggregates is available, it seems reasonable to assume that the formation of NMMHC-IIA inclusions derives from the presence in granulocytes of mutant molecules. In fact, the mutant NMMHC-IIA has intrinsic capability to form insoluble aggregates, as demonstrated for N93K by Hu et al. ( 30 ). In this context, the WT-mutant NMMHC-IIA interaction observed by Franke et al . ( 24 ) could result in the trapping of the WT molecules within the inclusion bodies. Non-muscle myosin II assembly involves at least two steps: dimerization of two alpha-helices to form coiled-coil rod structures and lateral association of coiled-coils to form functional filaments ( 14 ). Assuming that in granulocytes a random association of mutant and WT heavy chains occurs, 50% of myosin molecules should be heterodimers with one mutant and one WT heavy chain. The rest should be homodimers with two mutant (25%) or two WT (25%) chains. We suggest that our immunoblotting analyses of granulocyte NMMHC-IIA detected only the assemblies of WT chains, as the remaining molecules (homodimers of mutant chains and heterodimers of WT-mutant chains) were trapped within inclusion bodies and were lost at western blot assay. At the opposite, immunoblotting measured all the WT NMMHC-IIA of Mks because the absence of mutant protein prevented the formation of cytoplasmic aggregates. At ultrastructural analysis, granulocyte inclusions appear not membrane bound and contain an amorphous matrix with ribosomes and, sometimes, microfilaments. Different types of inclusions have been described so far, depending on shape, size, arrangement and periodicity of ribosomes, presence and orientation of microfilaments ( 31 – 34 ). Recent studies suggested an association between the specific- MYH9 mutations and the structure of granulocyte inclusions, which was observed both at immunofluorescence evaluation ( 10 ) and upon electron microscopy analyses ( 34 ). In this view, the different morphology of inclusions in patients with different mutations may reflect the distinct patterns of aberrant assembly observed by Franke et al . ( 24 ) by mixing different mutant NMMHC-IIA polypeptides with the WT forms. However, the constant presence of ribosomes suggests that the assembly of mutant and WT proteins into the inclusions always occurs during and/or shortly after transduction.
In conclusion, our study revealed for the first time that different MYH9 mutations lead to haploinsufficiency of NMMHC-IIA in Mks from patients with MYH9 -RD. This NMMHC-IIA deficiency does not prevent in vitro Mks differentiation, proliferation and maturation, and it is therefore likely to trigger a defective platelet production resulting in macrothrombocytopenia. At the opposite, we obtained evidences for a dominant-negative effect of mutant allele in granulocytes of the same patients showing haploinsufficiency in the megakaryocytic lineage. The reasons why the mutant NMMHC-IIA is differentially processed in Mks and granulocytes are still unknown. At the best of our knowledge, MYH9 -RD is the first example of a ‘dominant’ phenotype achieved by different processing of the same mutant molecule in different cell lineages, and further investigation on this topic is required for a better comprehension of this new mechanism of genetic disorders.
MATERIALS AND METHODS
Cases
Eleven patients from six unrelated MYH9 -RD pedigrees entered the study. MYH9 mutations and clinical and hematological features are summarized in Table 2 . Analysis of MGG-stained blood films revealed granulocyte Döhle-like inclusions in all the patients. All the cases presented characteristic NMMHC-IIA aggregates in granulocytes at immunofluorescence analysis with Abs against the head domain of NMMHC-IIA. Families B, C, D, E and F have been previously reported ( 3 , 9 ). Genotype analysis of family A was performed as described elsewhere ( 9 ). Twelve different healthy volunteers were studied as controls. Each subject gave written informed consent for the study.
Materials
Mouse monoclonal Ab NMF6, which recognizes an epitope of the head domain of platelet NMMHC-IIA ( 35 ), was a kind gift from Dr Saverio Sartore (Department of Biomedical Sciences, University of Padua, Italy). Rabbit polyclonal Ab PR-B440P was purchased from Covance Research Products (Berkeley, CA, USA). This Ab specifically recognizes a polypeptide sequence (GKADGAEAKPAE) corresponding to the COOH-terminus of human NMMHC-IIA ( 26 , 27 ). Monoclonal Abs AC-15 against β-actin, DM1A against α-tubulin, leupeptin, aprotinin and levamisole were from Sigma (St Louis, MI, USA). Bicinchoninic acid assay kit for protein determination and chemiluminescence substrates were from Pierce (Rockford, IL, USA). Peroxidase-conjugated goat anti-rabbit or anti-mouse Abs was from Bio-Rad (Richmond, CA, USA). Mouse monoclonal SZ21 anti-CD61 was from Immunotech (Marseille, France). Goat polyclonal anti-CD61 was from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Fluorochrome-conjugated secondary Abs and Hoechst 33258 penthahydrate were from Molecular Probes (Eugene, OR, USA).
Preparation of whole platelet lysates
Whole platelet lysates were obtained from all enrolled MYH9 -RD patients. PB samples from patients of each MYH9 -RD family were always processed simultaneously with one or two control samples. Twelve milliliter of PB from patients and controls were taken into citric acid/citrate/dextrose (ACD; 130 m m citric acid; 152 m m sodium citrate; 112 m m glucose) in a ratio of 9 to 1. Platelet-rich plasma was separated from whole blood by centrifugation at 100 g for 8 min. Platelets were then isolated by centrifugation at 700 g for 15 min in the presence of 0.2 U/ml apyrase grade I and 1 µ m PGE 1 and resuspended in PIPES buffer (136 m m NaCl, 20 m m PIPES, pH 6.5, 0.2 U/ml apyrase grade I, 1 µ m PGE 1 ). To prepare the whole platelet lysates, platelets were recovered by centrifugation at 2700 g for 3 min, solubilized with 100 µl of 2% SDS and boiled for 5 min.
Culture, separation and lysis of Mks
Liquid cultures of Mks from PB progenitors were carried out in all MYH9 -RD cases. Cultures from patients and controls were always performed simultaneously. Briefly, cultures were initiated by plating low-density (<1077 g/ml) cells obtained from 15 ml of PB in Stem Span Medium (Stem-Cell Technologies, Vancouver, Canada) containing 1% Penicillin–Streptomycin and 10 ng/ml thrombopoietin, IL6, IL11, Flt3-L (PeproTech EC Ltd, London, England). Cultures were maintained for 12 days at 37°C in a humidified atmosphere of 5% CO 2 . After 12 days, cultured cells were harvested and counted ( 36 ). CD41+ cells were separated by immunomagnetic bead technique (Miltenyi-Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. After collecting samples for microscopy analysis, the CD41+ fraction was recovered at 5300 g for 10 min and resuspended in 75 µl of ice-cold lysis buffer (10 m m Tris, 158 m m NaCl, 1% Triton X-100, 1% Na-deoxicholate, 0.1% SDS, 5 m m EGTA, pH 7.2, 1 m m PMSF, 1 m m NaVO 4 ) in the presence of 100 µg/ml leupeptin and 100 µg/ml aprotinin. Lysis was performed on ice for 20 min. Lysates were clarified by centrifugation at 15 700 g for 10 min at 4°C.
Results of Mk separation by immunomagnetic beads were evaluated by microscopy analysis after immunostaining for CD61 antigen. Mks were identified on the basis of CD61 expression plus cell morphology. A cell sample was considered suitable for immunoblotting analysis when Mks were >85% of total cells, without significant contamination with platelets (<3% of total cells). At least 2000 cells were observed for each specimen. Upon MGG staining, Mks were assigned to different stages of maturation according to standard morphological criteria ( 28 ).
To evaluate the Mk recovery at the end of liquid cultures, samples of total cultured cells were harvested at day 12 prior to Mk separation. Total cell counts were determined by an automated counter. Mk percentage was evaluated by microscopy analysis after immunoenzymatic staining for CD61. For each subject, at least 1×10 4 cells were observed by two-independent examinators. Analysis was performed in all the 11 MYH9 -RD patients and in 10 controls that were cultured in the same sets of experiments. Four patients were analyzed twice or three times (total, 17 assays). In these cases, median values were recorded together with median values of simultaneously cultured controls.
To investigate NMMHC-IIA distribution during in vitro Mk maturation, samples of total cultured cells were harvested at days 5, 7, 9 and 12. Aliquots of 5×10 4 cells were cytospun onto slides and double-stained for NMMHC-IIA and CD61 for immunofluorescence studies.
Separation and lysis of granulocytes
Granulocytes were separated from PB of seven patients from four MYH9 -RD families (A, B, C and E). Patient and control samples were always processed simultaneously. Twelve milliliter of PB was collected in a heparinized syringe. Supernatant obtained after erythrocyte sedimentation was centrifuged on a Ficoll-Hypaque gradient at 690 g for 45 min. The granulocyte pellet was recovered, resuspended in 80% NH 4 Cl 2 in PBS and incubated at 4°C for 20 min to obtain a complete lysis of residual red blood cells. Cells were then rinsed with PBS, recovered at 690 g for 10 min and resuspended in PBS. Removal of residual macrothrombocytes was obtained by collecting the negative fraction after separation of CD41+ cells by immunomagnetic bead technique (Miltenyi-Biotech). Cells were then centrifuged at 690 g for 10 min and resuspended in 150 µl of ice-cold lysis buffer plus 400 µg/ml leupeptin and 400 µg/ml aprotinin. Lysis was performed on ice for 20 min. Lysates were clarified by centrifugation at 15 700 g for 10 min at 4°C. Aliquots of 8×10 4 separated cells prior to lysis were cytospun onto slides for microscopy evaluation after MGG staining.
SDS–PAGE and immunoblotting
Upon evaluation of protein concentration, an equal volume of a mixture containing 1% dithiothreitol, 20% glycerol and 0.02% bromophenol blue was added to the platelet lysates. One-half volume of SDS sample buffer 3× (37.5 m m Tris, 288 m m glycine, pH 8.3, 6% SDS, 1.5% dithiothreitol, 30% glycerol, 0.03% bromophenol blue) was added to Mk and granulocyte lysates. Samples were then heated at 95°C for 5 min before being subjected to electrophoresis.
Identical amounts of proteins from each sample (20 µg for platelet and granulocyte lysates and 50 µg for Mks) were separated on 5–15% gradient acrylamide gels and transferred to nitrocellulose. Nitrocellulose membranes were then blocked overnight at 4°C with 6% BSA in 20 m m Tris/HCl, pH 7.5, 0.5 m NaCl and incubated with the primary Abs. The following Abs and dilutions were used: NMF6 anti-NMMHC-IIA, 1 : 1000; PRB440P anti-NMMHC-IIA, 1 : 1000; AC-15 anti-β-actin, 1 : 10000 and DM1A anti-α-tubulin, 1 : 1000. Upon incubation for 2 h at room temperature, membranes were extensively washed with buffer (50 m m Tris/HCl, pH 7.4, 0.2 m NaCl, 1 mg/ml polyethylene glycol 20000, 1% BSA, 0.05% Tween-20) and incubated with the appropriated peroxidase-conjugated secondary Ab, 1 : 3000, for 45 min. After thorough washing, reactive proteins were visualized with a chemiluminescence reaction.
Quantitative analysis of the intensity of the single bands was performed by densitometric scanning using a CAMAG TLC scanner. The NMMHC-IIA levels obtained by densitometric analysis were normalized upon β-actin levels and for each sample, the NMMHC-IIA/β-actin ratio was calculated to account for possible differences in protein loading. For each patient, the calculated NMMHC-IIA/β-actin ratio was expressed as the percentage of control conditions. Alpha-tubulin was used as further loading control, with results similar to those obtained with β-actin.
Immunostaining procedures, confocal and conventional microscopy
Immunoenzymatic staining for CD61 antigen was performed on cytospin preparations of cultured cells using the SZ21 Ab, 1:75. The Ab binding was revealed by an immunoalkaline-phosphatase method (LSAB ® 2 kit, Dako Corporation, Carpinteria, CA, USA). Levamisole 5 m m was added to the chromogen to inhibit endogenous enzymatic activity. Slides were counterstained with Mayer's Hemalum. Immunofluorescence double-labeling for NMMHC-IIA and CD61 was carried by using the NMF6 Ab, 1 : 300, in combination with goat polyclonal anti-CD61, 1 : 75. Secondary Abs were Alexa Fluor 594-conjugated chicken anti-mouse and Alexa Fluor 488-conjugated chicken anti-goat, both 1:200. Granulocytes were double-stained on PB smears by the NMF6, 1:300, in combination with the PR-B440P, 1:300. Slides were then incubated with the Alexa Fluor 488- or Alexa Fluor 594-conjugated appropriated goat Ab, 1:200. Hoechst 33258 penthahydrate, 100 ng/ml, was used for nuclear counterstaining. General staining procedures have been previously described ( 36 ). Negative controls were performed by replacing the primary Ab with an isotype-matched mouse monoclonal Ab or an irrelevant rabbit or goat polyclonal Ab.
Confocal microscopy was performed through the TCS SPII confocal laser scanning microscopy system (Leica, Heidelberg, Germany), equipped with a Leica DM IRBE inverted microscope. Analysis was carried out with a 63× immersion oil objective with 1.32 NA. The following laser line emissions were used: argon/visible 488 nm for Alexa Fluor 488, helium/neon 543 nm for Alexa Fluor 594 and argon/UV 364 nm for Hoechst. Band amplitude to detect fluorescence was comprised between 500 and 550 nm for Alexa Fluor 488, 590–670 nm for Alexa Fluor 594 and 400–500 nm for Hoechst. Images were acquired with a format of 1024×1024 pixels. Confocal optical sections were performed every 400 nm. Conventional microscopy was performed through an Axioscope 2 Plus microscope (Carl Zeiss, Gottingen, Germany), using a 63×/1.25 or a 100×/1.30 Plan Neofluar oil-immersion objective.
ACKNOWLEDGEMENTS
We thank Professor Saverio Sartore (Department of Medical Science, University of Padua, Italy) for providing the NMF6 antibody and Dr Patrizia Vaghi (Centro Grandi Strumenti, University of Pavia, Italy), for technical support with confocal microscopy. This work was supported by a grant from Italian Telethon Foundation (grant no. GP0019Y01).
Conflict of Interest statement. All authors have declared no conflict of interest.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
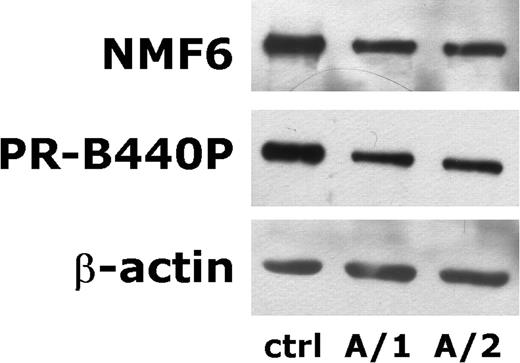
Figure 1. Representative immunoblots of platelet NMMHC-IIA. The figure reports a representative example of immunoblots obtained from platelet lysates of patients from family A. This family carries the R1933X mutation resulting in the deletion of a 27-residues COOH-terminal fragment of NMMHC-IIA. Twenty microgram of total platelet lysates were separated on a 5–15% gradient poliacrylamide gel, transferred to nitrocellulose and probed with an Ab recognizing an epitope of the head of NMMHC-IIA (NMF6), an Ab that specifically recognizes the last 12 COOH-terminal amino acids of NMMHC-IIA (PR-B440P) and with an anti-β-actin (AC-15), as indicated on the left. When compared with platelets from a healthy donor (ctrl), both patients from family A express a similar amount of β-actin, but a strongly reduced amount of NMMHC-IIA, as revealed by immunoblotting with NMF6 Ab. The figure also shows that the residual NMMHC-IIA expressed in platelets from patients A/1 and A/2 is the wild-type protein, as it reacts with the PR-B440P Ab whose epitope is deleted in the mutated protein.
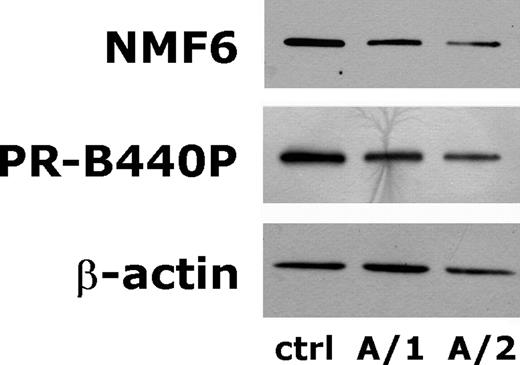
Figure 2. Representative immunoblots of megakaryocyte NMMHC-IIA. Megakaryocytes were cultured from PB progenitors of family A and purified by immunomagnetic beads technique on the basis of CD41 expression. Upon cell lysis, 50 µg of proteins were separated on polyacrylamide gel, transferred to nitrocellulose and probed with Abs NMF6 and PR-B440P anti-NMMHC-IIA and AC-15 anti-β-actin. While all the samples reported in the figure express similar amount of β-actin, analysis with the NMF6 Ab, which binds to both wild-type and mutant protein, or with Ab PR-B440P, which binds exclusively to the wild-type protein, revealed a reduced expression of NMMHC-IIA in Mks from both patients A/1 and A/2.
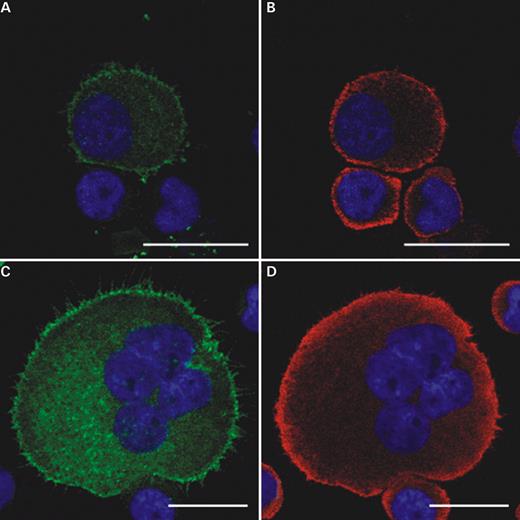
Figure 3. NMMHC-IIA distribution during megakaryocyte maturation. The figure reports a representative example of NMMHC-IIA distribution in one stage I megakaryocyte at day 5 of culture ( A and B ) and in one stage IV megakaryocyte at day 12 ( C and D ) obtained from patient A/2. Images correspond to confocal optical sections of cultured cells double-stained for CD61 (green) and NMMHC-IIA (red). In CD61+ cells, the NMMHC-IIA signal was organized in small dots diffusely distributed throughout the cytoplasm, with a reinforcement in the cortical region of the cell (B and D). Hoechst was used for nuclear counterstaining (blue). Scale bars correspond to 20 µm.
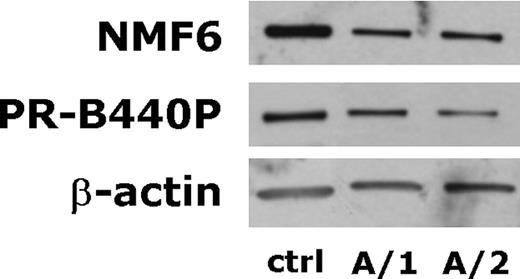
Figure 4. Representative immunoblots of granulocyte NMMHC-IIA. Granulocytes from family A were isolated from PB and finally lysed in ice-cold lysis buffer as described in Materials and Methods. Proteins were separated on a 5–15% acrylamide gel, transferred to nitrocellulose and probed with NMF6 and PR-B440P Abs anti-NMMHC-IIA and AC-15 Ab anti-β-actin. The figure shows an evident reduction of the expression of wild-type NMMHC-IIA in granulocytes from the representative patients A/1 and A/2. A similar reduction was detected with both the NMF6 Ab (recognizing both wild-type and mutant proteins) and PR-B440P Ab, whose epitope is deleted in the mutant NMMHC-IIA.
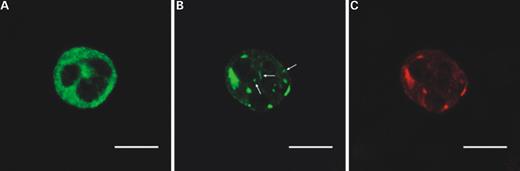
Figure 5. Granulocyte aggregates of NMMHC-IIA are stained by the PR-B440P antibody. Patient A/2, representative example of a granulocyte on PB smear double-stained with NMF6 Ab (green, B) and PR-B440P Ab (red, C). A representative example of a granulocyte from a healthy control stained with NMF6 Ab is shown in ( A ). In normal granulocytes, the NMMHC-IIA signal is homogeneously distributed within cell cytoplasm (A). In MYH9 -RD patients, immunofluorescence for NMMHC-IIA reveals characteristic cytoplasmic aggregates of the protein, corresponding to the sites of Döhle-like inclusions, with a faint residual diffuse staining in the cytoplasm. The figure outlines that most of the aggregates of NMMHC-IIA recognized by the NMF6 Ab against the head of the molecule ( B ) are stained also by the PR-B440P Ab against a COOH-terminal fragment deleted in mutant NMMHC-IIA ( C ), indicating that the WT NMMHC-IIA is sequestered in these aggregates. Only few aggregates with small size (arrow) are recognized exclusively by the NMF6 Ab. Scale bars correspond to 10 µm.
Results of densitometric analysis of immunoblottings
| Family/patient | Platelets N/A ratio versus controls (%) | Megakaryocytes N/A ratio versus controls (%) | Granulocytes N/A ratio versus controls (%) | |||
|---|---|---|---|---|---|---|
| NMF6 Ab | PR-B440P Ab | NMF6 Ab | PR-B440P Ab | NMF6 Ab | PR-B440P Ab | |
| A/1 | 50 | 47 | 48 | 44 | 30 | 41 |
| A/2 | 49 | 48 | 42 | 40 | 32 | 21 |
| B/1 | 65 | 47 | 44 | 40 | 34 | 42 |
| B/2 | 36 | 32 | 42 | 42 | 28 | 26 |
| B/3 | 50 | 58 | 45 | 49 | 18 | 18 |
| C/1 | 55 | nd | 54 | |||
| D/1 | 58 | nd | nd | |||
| D/2 | 36 | nd | nd | |||
| E/1 | 50 | nd | 36 | |||
| F/1 | 68 | 49 | nd | |||
| F/2 | 41 | 54 | nd | |||
| Median | 50 | 47 | 45 | 42 | 32 | 26 |
| Mean | 49.0 | 46.4 | 46.2 | 43.0 | 33.1 | 29.6 |
| SD | 9.3 | 9.3 | 4.3 | 3.7 | 10.9 | 11.2 |
| Family/patient | Platelets N/A ratio versus controls (%) | Megakaryocytes N/A ratio versus controls (%) | Granulocytes N/A ratio versus controls (%) | |||
|---|---|---|---|---|---|---|
| NMF6 Ab | PR-B440P Ab | NMF6 Ab | PR-B440P Ab | NMF6 Ab | PR-B440P Ab | |
| A/1 | 50 | 47 | 48 | 44 | 30 | 41 |
| A/2 | 49 | 48 | 42 | 40 | 32 | 21 |
| B/1 | 65 | 47 | 44 | 40 | 34 | 42 |
| B/2 | 36 | 32 | 42 | 42 | 28 | 26 |
| B/3 | 50 | 58 | 45 | 49 | 18 | 18 |
| C/1 | 55 | nd | 54 | |||
| D/1 | 58 | nd | nd | |||
| D/2 | 36 | nd | nd | |||
| E/1 | 50 | nd | 36 | |||
| F/1 | 68 | 49 | nd | |||
| F/2 | 41 | 54 | nd | |||
| Median | 50 | 47 | 45 | 42 | 32 | 26 |
| Mean | 49.0 | 46.4 | 46.2 | 43.0 | 33.1 | 29.6 |
| SD | 9.3 | 9.3 | 4.3 | 3.7 | 10.9 | 11.2 |
For each sample, the NMMHC-IIA levels obtained by densitometric scanning were normalized upon β-actin levels, and the NMMHC-IIA/β-actin ratio (N/A ratio) was calculated to account for differences in protein loading. The values of N/A ratio were expressed as the percentages of respective control conditions for each cell type. nd, not done.
Results of densitometric analysis of immunoblottings
| Family/patient | Platelets N/A ratio versus controls (%) | Megakaryocytes N/A ratio versus controls (%) | Granulocytes N/A ratio versus controls (%) | |||
|---|---|---|---|---|---|---|
| NMF6 Ab | PR-B440P Ab | NMF6 Ab | PR-B440P Ab | NMF6 Ab | PR-B440P Ab | |
| A/1 | 50 | 47 | 48 | 44 | 30 | 41 |
| A/2 | 49 | 48 | 42 | 40 | 32 | 21 |
| B/1 | 65 | 47 | 44 | 40 | 34 | 42 |
| B/2 | 36 | 32 | 42 | 42 | 28 | 26 |
| B/3 | 50 | 58 | 45 | 49 | 18 | 18 |
| C/1 | 55 | nd | 54 | |||
| D/1 | 58 | nd | nd | |||
| D/2 | 36 | nd | nd | |||
| E/1 | 50 | nd | 36 | |||
| F/1 | 68 | 49 | nd | |||
| F/2 | 41 | 54 | nd | |||
| Median | 50 | 47 | 45 | 42 | 32 | 26 |
| Mean | 49.0 | 46.4 | 46.2 | 43.0 | 33.1 | 29.6 |
| SD | 9.3 | 9.3 | 4.3 | 3.7 | 10.9 | 11.2 |
| Family/patient | Platelets N/A ratio versus controls (%) | Megakaryocytes N/A ratio versus controls (%) | Granulocytes N/A ratio versus controls (%) | |||
|---|---|---|---|---|---|---|
| NMF6 Ab | PR-B440P Ab | NMF6 Ab | PR-B440P Ab | NMF6 Ab | PR-B440P Ab | |
| A/1 | 50 | 47 | 48 | 44 | 30 | 41 |
| A/2 | 49 | 48 | 42 | 40 | 32 | 21 |
| B/1 | 65 | 47 | 44 | 40 | 34 | 42 |
| B/2 | 36 | 32 | 42 | 42 | 28 | 26 |
| B/3 | 50 | 58 | 45 | 49 | 18 | 18 |
| C/1 | 55 | nd | 54 | |||
| D/1 | 58 | nd | nd | |||
| D/2 | 36 | nd | nd | |||
| E/1 | 50 | nd | 36 | |||
| F/1 | 68 | 49 | nd | |||
| F/2 | 41 | 54 | nd | |||
| Median | 50 | 47 | 45 | 42 | 32 | 26 |
| Mean | 49.0 | 46.4 | 46.2 | 43.0 | 33.1 | 29.6 |
| SD | 9.3 | 9.3 | 4.3 | 3.7 | 10.9 | 11.2 |
For each sample, the NMMHC-IIA levels obtained by densitometric scanning were normalized upon β-actin levels, and the NMMHC-IIA/β-actin ratio (N/A ratio) was calculated to account for differences in protein loading. The values of N/A ratio were expressed as the percentages of respective control conditions for each cell type. nd, not done.
Clinical and laboratory features of the 11 investigated MYH9 -RD patients
| Family/patient (ref) | Sex/age | MYH9 mutation | Platelet count a (×10 9 /l) | Distribution of platelet diameters b (%) | WBC count (×10 9 /l) | Hearing impairment c | Kidney impairment | Cataracts d | ||
|---|---|---|---|---|---|---|---|---|---|---|
| <4 µm | 4–8 µm | >8 µm | ||||||||
| A/1 | F/59 | R1933X | 24 | 29.2 | 45.1 | 25.7 | 5.5 | B-SNHLHT | No | No |
| A/2 | F/33 | R1933X | 50 | 35.0 | 49.1 | 15.9 | 5.9 | No | No | No |
| B/1 ( 9 ) | F/56 | E1945X | 63 | 60.0 | 32.0 | 8.0 | 5.9 | B-SNHLHT | Proteinuria, microscopic haematuria | No |
| B/2 ( 9 ) | M/30 | E1945X | 128 | 70.3 | 20.5 | 9.2 | 9.5 | No | Proteinuria, microscopic haematuria | No |
| B/3 ( 9 ) | F/29 | E1945X | 68 | 69.7 | 21.7 | 8.6 | 12.8 | No | Proteinuria, microscopic haematuria | No |
| C/1 ( 3 ) | F/44 | R1165C | 88 | 63.1 | 27.9 | 9.0 | 9.2 | B-SNHLHT | No | No |
| D/1 ( 9 ) | F/60 | D1424 N | 55 | 60.8 | 26.6 | 12.6 | 5.0 | B-SNHLHT | No | No |
| D/2 ( 9 ) | F/37 | D1424 N | 30 | 62.5 | 24.0 | 13.5 | 5.1 | No | No | No |
| E/1 ( 3 ) | F/39 | N93K | 26 | 20.9 | 35.1 | 43.9 | 7.7 | B-SNHLHT | Proteinuria, microscopic haematuria | No |
| F/1 ( 3 ) | M/57 | E1841K | 82 | 68.5 | 24.1 | 7.4 | 5.4 | B-SNHLHT | No | Bilateral (38) |
| F/2 ( 3 ) | M/16 | E1841K | 129 | 69.4 | 21.8 | 8.8 | 5.0 | No | No | No |
| Family/patient (ref) | Sex/age | MYH9 mutation | Platelet count a (×10 9 /l) | Distribution of platelet diameters b (%) | WBC count (×10 9 /l) | Hearing impairment c | Kidney impairment | Cataracts d | ||
|---|---|---|---|---|---|---|---|---|---|---|
| <4 µm | 4–8 µm | >8 µm | ||||||||
| A/1 | F/59 | R1933X | 24 | 29.2 | 45.1 | 25.7 | 5.5 | B-SNHLHT | No | No |
| A/2 | F/33 | R1933X | 50 | 35.0 | 49.1 | 15.9 | 5.9 | No | No | No |
| B/1 ( 9 ) | F/56 | E1945X | 63 | 60.0 | 32.0 | 8.0 | 5.9 | B-SNHLHT | Proteinuria, microscopic haematuria | No |
| B/2 ( 9 ) | M/30 | E1945X | 128 | 70.3 | 20.5 | 9.2 | 9.5 | No | Proteinuria, microscopic haematuria | No |
| B/3 ( 9 ) | F/29 | E1945X | 68 | 69.7 | 21.7 | 8.6 | 12.8 | No | Proteinuria, microscopic haematuria | No |
| C/1 ( 3 ) | F/44 | R1165C | 88 | 63.1 | 27.9 | 9.0 | 9.2 | B-SNHLHT | No | No |
| D/1 ( 9 ) | F/60 | D1424 N | 55 | 60.8 | 26.6 | 12.6 | 5.0 | B-SNHLHT | No | No |
| D/2 ( 9 ) | F/37 | D1424 N | 30 | 62.5 | 24.0 | 13.5 | 5.1 | No | No | No |
| E/1 ( 3 ) | F/39 | N93K | 26 | 20.9 | 35.1 | 43.9 | 7.7 | B-SNHLHT | Proteinuria, microscopic haematuria | No |
| F/1 ( 3 ) | M/57 | E1841K | 82 | 68.5 | 24.1 | 7.4 | 5.4 | B-SNHLHT | No | Bilateral (38) |
| F/2 ( 3 ) | M/16 | E1841K | 129 | 69.4 | 21.8 | 8.8 | 5.0 | No | No | No |
ref, reference. M- and B-SNHLHT: mono- and bi-lateral sensorineural hearing loss for high tones. nd, not determined.
a As determined by phase contrast microscopy.
b As determined by analysis of blood smears prepared without anticoagulant and stained by MGG. Normal values: <4 µm: 87–100%, 4–8 µm: 0–10%, >8 µm: 0–1%, as determined by analysis of 50 healthy subjects.
c As determined by audiogram.
d In parentheses is reported the age of onset.
Clinical and laboratory features of the 11 investigated MYH9 -RD patients
| Family/patient (ref) | Sex/age | MYH9 mutation | Platelet count a (×10 9 /l) | Distribution of platelet diameters b (%) | WBC count (×10 9 /l) | Hearing impairment c | Kidney impairment | Cataracts d | ||
|---|---|---|---|---|---|---|---|---|---|---|
| <4 µm | 4–8 µm | >8 µm | ||||||||
| A/1 | F/59 | R1933X | 24 | 29.2 | 45.1 | 25.7 | 5.5 | B-SNHLHT | No | No |
| A/2 | F/33 | R1933X | 50 | 35.0 | 49.1 | 15.9 | 5.9 | No | No | No |
| B/1 ( 9 ) | F/56 | E1945X | 63 | 60.0 | 32.0 | 8.0 | 5.9 | B-SNHLHT | Proteinuria, microscopic haematuria | No |
| B/2 ( 9 ) | M/30 | E1945X | 128 | 70.3 | 20.5 | 9.2 | 9.5 | No | Proteinuria, microscopic haematuria | No |
| B/3 ( 9 ) | F/29 | E1945X | 68 | 69.7 | 21.7 | 8.6 | 12.8 | No | Proteinuria, microscopic haematuria | No |
| C/1 ( 3 ) | F/44 | R1165C | 88 | 63.1 | 27.9 | 9.0 | 9.2 | B-SNHLHT | No | No |
| D/1 ( 9 ) | F/60 | D1424 N | 55 | 60.8 | 26.6 | 12.6 | 5.0 | B-SNHLHT | No | No |
| D/2 ( 9 ) | F/37 | D1424 N | 30 | 62.5 | 24.0 | 13.5 | 5.1 | No | No | No |
| E/1 ( 3 ) | F/39 | N93K | 26 | 20.9 | 35.1 | 43.9 | 7.7 | B-SNHLHT | Proteinuria, microscopic haematuria | No |
| F/1 ( 3 ) | M/57 | E1841K | 82 | 68.5 | 24.1 | 7.4 | 5.4 | B-SNHLHT | No | Bilateral (38) |
| F/2 ( 3 ) | M/16 | E1841K | 129 | 69.4 | 21.8 | 8.8 | 5.0 | No | No | No |
| Family/patient (ref) | Sex/age | MYH9 mutation | Platelet count a (×10 9 /l) | Distribution of platelet diameters b (%) | WBC count (×10 9 /l) | Hearing impairment c | Kidney impairment | Cataracts d | ||
|---|---|---|---|---|---|---|---|---|---|---|
| <4 µm | 4–8 µm | >8 µm | ||||||||
| A/1 | F/59 | R1933X | 24 | 29.2 | 45.1 | 25.7 | 5.5 | B-SNHLHT | No | No |
| A/2 | F/33 | R1933X | 50 | 35.0 | 49.1 | 15.9 | 5.9 | No | No | No |
| B/1 ( 9 ) | F/56 | E1945X | 63 | 60.0 | 32.0 | 8.0 | 5.9 | B-SNHLHT | Proteinuria, microscopic haematuria | No |
| B/2 ( 9 ) | M/30 | E1945X | 128 | 70.3 | 20.5 | 9.2 | 9.5 | No | Proteinuria, microscopic haematuria | No |
| B/3 ( 9 ) | F/29 | E1945X | 68 | 69.7 | 21.7 | 8.6 | 12.8 | No | Proteinuria, microscopic haematuria | No |
| C/1 ( 3 ) | F/44 | R1165C | 88 | 63.1 | 27.9 | 9.0 | 9.2 | B-SNHLHT | No | No |
| D/1 ( 9 ) | F/60 | D1424 N | 55 | 60.8 | 26.6 | 12.6 | 5.0 | B-SNHLHT | No | No |
| D/2 ( 9 ) | F/37 | D1424 N | 30 | 62.5 | 24.0 | 13.5 | 5.1 | No | No | No |
| E/1 ( 3 ) | F/39 | N93K | 26 | 20.9 | 35.1 | 43.9 | 7.7 | B-SNHLHT | Proteinuria, microscopic haematuria | No |
| F/1 ( 3 ) | M/57 | E1841K | 82 | 68.5 | 24.1 | 7.4 | 5.4 | B-SNHLHT | No | Bilateral (38) |
| F/2 ( 3 ) | M/16 | E1841K | 129 | 69.4 | 21.8 | 8.8 | 5.0 | No | No | No |
ref, reference. M- and B-SNHLHT: mono- and bi-lateral sensorineural hearing loss for high tones. nd, not determined.
a As determined by phase contrast microscopy.
b As determined by analysis of blood smears prepared without anticoagulant and stained by MGG. Normal values: <4 µm: 87–100%, 4–8 µm: 0–10%, >8 µm: 0–1%, as determined by analysis of 50 healthy subjects.
c As determined by audiogram.
d In parentheses is reported the age of onset.
References
Balduini, C.L. and Savoia, A. (
Geddis, A.E. and Kaushansky, K. (
Seri, M., Cusano, R., Gangarossa, S., Caridi, G., Bordo, D., Lo Nigro, C., Ghiggeri, G.M., Ravazzolo, R., Savino, M., Del Vecchio, M. et al . (
Kelley, M.J., Jawien, W., Ortel, T.L. and Korczak, J.F. (
Kunishima, S., Kojima, T., Matsushita, T., Tanaka, T., Tsurusawa, M., Furukawa, Y., Nakamura, Y., Okamura, T., Amemiya, N., Nakayama, T. et al . (
Heath, K.E., Campos-Barros, A., Toren, A., Rozenfeld-Granot, G., Carlsson, L.E., Savige, J., Denison, J.C., Gregory, M.C., White, J.G., Barker, D.F. et al . (
Arrondel, C., Vodovar, N., Knebelmann, B., Grunfeld, J.P., Gubler, M.C., Antignac, C. and Heidet, L. (
Seri, M., Savino, M., Bordo, D., Cusano, R., Rocca, B., Meloni, I., Di Bari, F., Koivisto, P.A., Bolognesi, M., Ghiggeri, G.M. et al. (
Seri, M., Pecci, A., Di Bari, F., Cusano, R., Savino, M., Panza, E., Del Vecchio, M., Noris, P., Gangarossa, S., Rocca, B. et al. (
Kunishima, S., Matsushita, T., Kojima, T., Sako, M., Kimura, F., Jo, E.K., Inoue, C., Kamiya, T. and Saito, H. (
Kunishima, S., Matsushita, T., Kojima, T., Amemiya, N., Choi, Y.M., Hosaka, N., Inoue, M., Jung, Y., Mamiya, S., Matsumoto, K. et al . (
Marigo, V., Nigro, A., Pecci, A., Montanaro, D., Di Stazio, M.T., Balduini, C.L. and Savoia, A. (
Robinson, D.N. and Spudich, J.A. (
Wei, Q. and Adelstein, R.S. (
Straight, A.F., Cheung, A., Limouze, J., Chen, I., Westwood, N.J., Sellers, J.R. and Mitchinson, T.J. (
Jacobelli, J., Chmura, S.A., Buxton, D.B., Davis, M.M. and Krummel, M.F. (
Ikebe, M., Komatsu, S., Woodhead, J.L., Mabuchi, K., Ikebe, R., Saito, J., Craig, R. and Higashihara, M. (
Balduini, C.L., Iolascon, A. and Savoia, A. (
Godwin, H.A. and Ginsburg, A.D. (
Hamilton, R.W., Shalk, B.S., Ottie, J.N., Storch, A.E., Saleem, A. and White, J.G. (
Heynen, M.J., Blockmans, D., Verwilghen, R.L. and Vermylen, J. (
Ghiggeri, G.M., Caridi, G., Magrini, U., Sessa, A., Savoia, A., Seri, M., Pecci, A., Romagnoli, R., Gangarossa, S., Noris, P. et al . (
Franke J.D., Dong, F., Rickoll, W.L., Kelley, M.J. and Kiehart, D.P. (
Deutsch, S., Rideau, A., Bochaton-Piallat, M.L., Merla, G., Geinoz, A., Gabbiani, G., Schwede, T., Matthes, T., Antonarakis, S.E. and Beris, P. (
Maupin, P., Phillips, C.L., Adelstein, R.S. and Pollard, T.D. (
Phillips, C.L., Yamakawa, K. and Adelstein, R.S. (
Williams, N. and Levine, R.F. (
Noris, P., Spedini, P., Belletti, S., Magrini, U. and Balduini, C.L. (
Hu, A., Wang, F. and Sellers, J.R. (
Greinacher, A. and Mueller-Eckhardt, C. (
Pujol-Moix, N., Muniz-Diaz, E., Moreno-Torres, M.L., Hernandez, A., Romero, M.A. and Puig, J. (
White, J.G., Mattson, J.C., Nichols, W.L., Luban, N.L.C. and Greinacher, A. (
Pujol-Moix, N., Kelley, M.J., Hernandez, A., Muniz-Diaz, E. and Espanol, I. (
Frid, M.G., Printesva, O.Y., Chiavegato, A., Faggin, E., Scatena, M., Koteliansky, V.E., Pauletto, P., Glukhova, M.A. and Sartore, S. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}