




Abstract
Mutations in the gene encoding glutamine-fructose-6-phosphate transaminase 1 (GFPT1) cause the neuromuscular disorder limb-girdle congenital myasthenic syndrome (LG-CMS). One recurrent GFPT1 mutation detected in LG-CMS patients is a c.*22C>A transversion in the 3′-untranslated region (UTR). Because this variant does not alter the GFPT1 open reading frame, its pathogenic relevance has not yet been established. We found that GFPT1 protein levels were reduced in myoblast cells of the patients carrying this variant. In silico algorithms predicted that the mutation creates a microRNA target site for miR-206*. Investigation of the expression of this so far unrecognized microRNA confirmed that miR-206* (like its counterpart miR-206) is abundant in skeletal muscle. MiR-206* efficiently reduced the expression of reporter constructs containing the mutated 3′-UTR while no such effect was observed with reporter constructs containing the wild-type 3′-UTR or when a specific anti-miR-206* inhibitor was added. Moreover, anti-miR-206* inhibitor treatment substantially rescued GFPT1 expression levels in patient-derived myoblasts. Our data demonstrate that the c.*22C>A mutation in the GFPT1 gene leads to illegitimate binding of microRNA resulting in reduced protein expression. We confirm that c.*22C>A is a causative mutation and suggest that formation of microRNA target sites might be a relevant pathomechanism in Mendelian disorders. Variants in the 3′-UTRs should be considered in genetic diagnostic procedures.
Introduction
Congenital myasthenic syndromes (CMS) are inherited disorders characterized by defects in neuromuscular transmission resulting from mutations in a number of different genes. CMS are clinically similar to the autoimmune disorder myasthenia gravis, lead to transient pareses and, if untreated, may also cause generalized paralysis and respiratory failure (1,2). Autosomal recessive mutations in the gene encoding glutamine-fructose-6-phosphate transaminase 1 (GFPT1) were identified in CMS patients with prominent limb-girdle weakness, frequent tubular aggregates in skeletal muscle and good response to acetylcholinesterase inhibitor therapy (3). GFPT1 is the first and rate-limiting enzyme of the hexosamine biosynthetic pathway leading to UDP-N-acetylglucosamine (UDP-GlcNAc). UDP-GlcNAc serves as precursor for amino sugars that are essential for protein and lipid modifications (4,5). As we have previously shown (3) and has been confirmed by others (6), GFPT1 missense mutations generally lead to reduced GFPT1 protein levels in patient muscle biopsies and cultured muscle cells. One of the GFPT1 mutations identified in four independent families from Spain, Germany and France was the 3′-untranslated region (UTR) mutation c.*22C>A (3,7). In all four families the mutation was compound heterozygous to missense or protein truncating mutations. The pathogenic relevance of the mutation c.*22C>A has not yet been supported by functional studies and the mechanism how this mutation impairs GFPT1 function has remained unclear.
The 3′-UTR of genes has various regulatory functions including determination of messenger RNA (mRNA) stability/degradation, subcellular localization, nuclear export and translation efficiency (8–10). A class of small non-coding RNA molecules (microRNAs) bind to the 3′-UTRs of target mRNAs and mediate translational repression through the degradation of the mRNA or translational inhibition (11). Primary microRNAs (pri-microRNAs) are transcribed by polymerase II or III, processed to precursor microRNA (pre-microRNA) hairpins and transported to the cytoplasm. Once arrived, they are cleaved by Dicer, resulting in microRNA duplexes of about 21–23 nucleotides (nt) and the strands are selectively loaded into the RNA-induced silencing complex (RISC) (11,12). Strand selection correlates with the thermodynamic stability of each end of the duplex (13–15). The more abundant strand is conventionally called microRNA, whereas its less abundant counterpart is known as the star-form microRNA (microRNA*) (12,16). However, recent studies revealed that some microRNA* species are well expressed and that the star-form may also have important regulatory functions (16–18). In addition, microRNA/microRNA* ratios seem to vary dramatically between developmental stages (14,19,20).
The critical role of microRNAs in skeletal muscle has recently emerged from conditional inactivation of Dicer in embryonic skeletal muscle which resulted in skeletal muscle hypoplasia, abnormal myofiber morphology and perinatal death (21). Several microRNAs that contribute to skeletal myogenesis have been identified and characterized (22,23). The best studied examples are miR-1, miR-133a7b and miR-206 (also called MyomiRs (24)). Because microRNA binding to 3′-UTRs can suppress protein expression, we investigated whether the GFPT1 3′-UTR mutation c.*22C>A may create an illegitimate microRNA binding site.
Results
GFPT1 protein expression and quantification of GFPT1 mRNA in muscle cells harbouring the c.*22C>A mutation
Western blot analysis of myoblasts obtained from three GFPT1 patients carrying the c.*22C>A mutation (and a missense change on the second allele) revealed reduced GFPT1 protein expression (Fig. 1A). Thus we confirmed our previous findings that this variant in the 3′-UTR of GFPT1 is associated with reduced amounts of GFPT1 protein (3). Based on cDNA sequencing data we had also suggested earlier that similar amounts of the mutated mRNA are present relative to the mRNA transcribed from the second allele (3). To corroborate these preliminary data we quantified GFPT1 mRNA levels in total RNA derived from myoblasts of patients with the c.*22C>A mutation by real-time qRT-PCR. No gross changes in the GFPT1 mRNA level were observed compared with control individuals (Fig. 1B). Altogether, these results indicate that the reduced amounts of GFPT1 protein levels in myoblasts of patients with the c.*22C>A GFPT1 mutation result from repression of translation rather than altered mRNA stability.
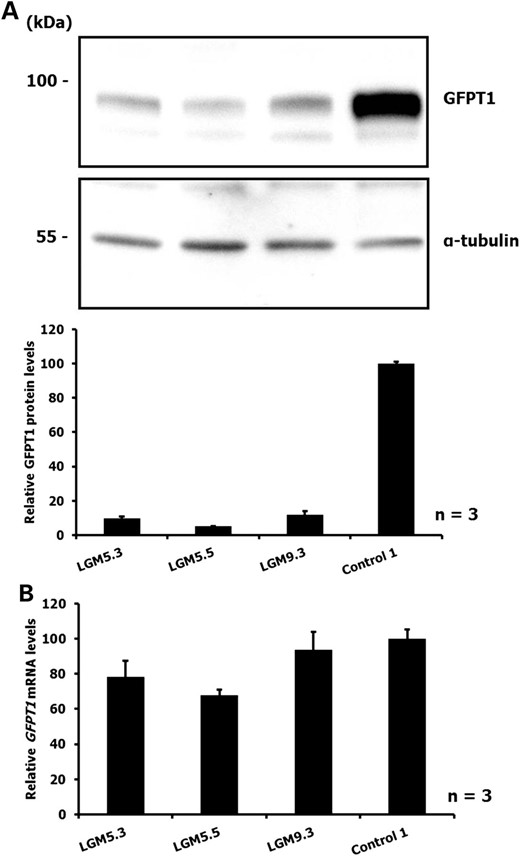
(A) Immunoblot detection of GFPT1 in myoblasts of patients with the GFPT1 mutation c.*22C>A and heteroallelic missense mutations (LGM5.3 and LGM5.5: p.M492T; LGM9.3: p.V199F). Control 1: healthy control individual. Upper panel: representative result from three independent experiments. Lower panel: The intensities of the bands were measured and GFPT1 expression was normalized to α-tubulin levels. Data are shown relative to the GFPT1 level of the control individual. (B) Levels of GFPT1 mRNA in myoblasts obtained from GFPT1 patients compound heterozygous with the c.*22C>A mutation (LGM5.3, LGM5.5 and LGM9.3; for missense mutations on the second allele, see (A)). Control 1: healthy control individual. Transcript levels were analysed by real-time qRT-PCR and normalized to histone hH4 levels. Data represent three independent experiments and are shown relative to the GFPT1 level of the control individual.
Expression analysis of the 3′-UTR mutation c.*22C>A
Because patients with the c.*22C>A mutation carry a GFPT1 missense change on the other allele reduced GFPT1 protein amounts in myoblasts reflect the impact of both mutations. Therefore, the effect of the c.*22C>A mutation on GFPT1 protein expression was further analysed in a controlled experiment in C2C12 myoblasts transfected with expression vectors containing either wild-type GFPT1 or mutant c.*22C>A GFPT1. Immunoblot analysis revealed that the 3′-UTR mutation leads to a significant decrease in GFPT1 expression compared with wild-type (Fig. 2) confirming the association of GFPT1 c.*22C>A with reduced GFPT1 protein levels. Real-time qRT-PCR confirmed that levels of GFPT1 mRNA expressed from both constructs were similar (Fig. 2B).
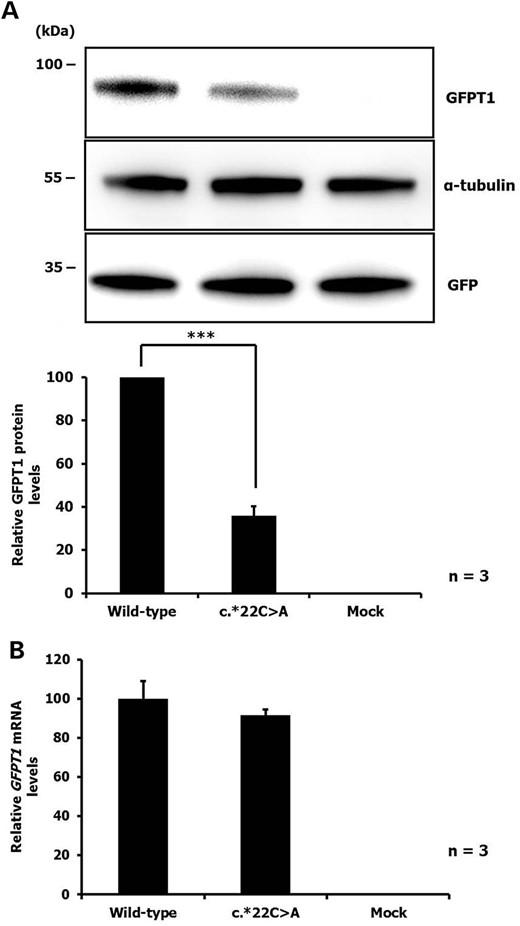
(A) Immunoblot of GFPT1 in C2C12 cells transiently transfected with GFPT1-3′-UTR wild-type or mutant (c.*22C>A) constructs. The cells were co-transfected with a GFP expression vector to control for transfection efficiency. Upper panel: representative result from three independent experiments. Lower panel: The intensities of the bands were measured and GFPT1 expression was normalized to the GFP and α-tubulin levels. Bars represent the average GFPT1 protein levels relative to the wild-type GFPT1 level. P < 0.001, wild-type versus c.22C>A. (B) Relative levels of GFPT1 mRNA in C2C12 cells transiently transfected with GFPT1-3′-UTR wild-type or mutant (c.*22C>A) constructs. Transcript levels were analysed by real-time qRT-PCR and normalized to mGAPDH levels. Data represent three independent experiments and are shown relative to the wild-type GFPT1 level.
The c.*22C>A mutation and microRNA binding sites in the GFPT1 3′-UTR
We next asked how the c.*22C>A mutation might influence GFPT1 protein expression. Because one mechanism of expression regulation is microRNA binding to 3′-UTR regions of target mRNAs, we performed in silico analyses to detect potential microRNA binding sites. The c.*22C>A variant was predicted to lead to the gain of putative binding sites for miR-206* and miR-600. Although there is no perfect seed matching (position 2–7 of the mature microRNA), the c.*22C>A variant strengthens a potential compensatory base pairing site in the 3′-region of the miR-206* by contributing an additional Watson–Crick base pairing (A:U) just before the anchor (nucleotides 13–16) (Fig. 3A). Furthermore, the in silico analysis revealed that the c.*22C>A variant resides inside a sequence matching the seed of mature miR-600 (5′-CUUACA-3′; Supplementary Material, Fig. S1).
![(A) Schematic representation of the sequence alignment of the miR-206* with wild-type and the variant (c.*22C>A) GFPT1 mRNA. Bioinformatics tools revealed that the 3′-UTR mutation c.*22C>A may result in a gain of a putative binding site for the microRNA miR-206* [MIMAT0006994]. The miR-206* alignment shows imperfect seed pairing, but compensatory pairing in the 3′-region of the microRNA. The GFPT1 3′-UTR c.*22C>A mutation is shown in red and contributes an additional Watson–Crick base pairing which is predicted to stabilize miR-206* binding. The seed region is highlighted in grey. (B) Detection of miR-206 and miR-206* in primary human myoblasts, myotubes and muscle biopsies by RT-PCR. U6 snRNA was used as an internal control. Controls are from healthy individuals; myoblasts are of patient LGM9.3: compound heterozygous for c.*22C>A and p.V199F; muscle biopsy is of patient LGM71: compound heterozygous for c.*22C>A and p.A550V; note that expression of the miRNAs in myotubes of the patient could not be analysed because patient cells did not differentiate into myotubes. (C) Quantitative analysis of the microRNAs miR-206 and miR-206* in myoblasts and myotubes of a healthy donor (Control 1) by real-time qRT-PCR. Error bars indicate +SD obtained from three replicates.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/24/12/10.1093/hmg/ddv090/2/m_ddv09003.jpeg?Expires=1716404762&Signature=T1G7EBvS6pwcY3ILQRURrF861PCcyMC~9UozzRl2acFAyjbGA4JH06eF2VjYNft~2iP~fWuvI8YWxKODraZjIati-1fmV3Ry2~AFO-nTl5b0tnbAWYL1EK9UT7zxcFa1wOSs9EsAUp6ztVs57cYn-WJQROEodoxju-UzxHolwSLraeh6gtECcUc0o0NwwUzyeulLKpjRLfdcDofAnxTCEDdU~zVEd9atIM2tPtuARuSLBU728wEAH7UuxD1Jmxv5Pv4DP~s5tgx2kkwhSoBO5Cl24bjWGcu6y3w5mm1H6PKC0yhHY~1kNAPNfVe6Lu5mJqPw1-2M5TvoOiuJBmlpIQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(A) Schematic representation of the sequence alignment of the miR-206* with wild-type and the variant (c.*22C>A) GFPT1 mRNA. Bioinformatics tools revealed that the 3′-UTR mutation c.*22C>A may result in a gain of a putative binding site for the microRNA miR-206* [MIMAT0006994]. The miR-206* alignment shows imperfect seed pairing, but compensatory pairing in the 3′-region of the microRNA. The GFPT1 3′-UTR c.*22C>A mutation is shown in red and contributes an additional Watson–Crick base pairing which is predicted to stabilize miR-206* binding. The seed region is highlighted in grey. (B) Detection of miR-206 and miR-206* in primary human myoblasts, myotubes and muscle biopsies by RT-PCR. U6 snRNA was used as an internal control. Controls are from healthy individuals; myoblasts are of patient LGM9.3: compound heterozygous for c.*22C>A and p.V199F; muscle biopsy is of patient LGM71: compound heterozygous for c.*22C>A and p.A550V; note that expression of the miRNAs in myotubes of the patient could not be analysed because patient cells did not differentiate into myotubes. (C) Quantitative analysis of the microRNAs miR-206 and miR-206* in myoblasts and myotubes of a healthy donor (Control 1) by real-time qRT-PCR. Error bars indicate +SD obtained from three replicates.
Expression profile of the microRNAs
As the expression of microRNAs is spatially and temporally controlled, we investigated the expression profile of miR-206, miR-206* and miR-600. We confirmed by RT-PCR that miR-206 is present in myoblasts, myotubes and skeletal muscle and found that miR-206* is robustly expressed in muscle and myoblast samples of controls and GFPT1 patients as well (Fig. 3B). It has been shown that miR-206 is induced during the myoblast–myotube transition (25,26). Consistent with this, real-time qRT-PCR assays showed that miR-206* is also upregulated upon myoblast differentiation (Fig. 3C). Furthermore, we found that both miR-206 and miR-206* are more abundant in myoblasts of GFPT1 patients than in control individuals (Supplementary Material, Fig. S2). We failed to develop conditions that allow for specific detection of expression of miR-600 in cultured skeletal muscle cells. RT-PCR yielded variable additional bands precluding precise measurements of miR-600 levels.
Reporter assay testing the interaction between miR-206* and GFPT1 mRNA
In order to test the hypothesis that the GFPT1 3′-UTR mutation c.*22C>A creates a novel miR-206* binding site, a reporter gene assay was performed. Either four tandem repeats (to increase the effect) or a single copy of an 80 bp sequence around GFPT1 c.*22 with either C (wild-type) or A (c.*22C>A) was subcloned downstream of the luciferase gene of pRL-TK (Fig. 4A). In agreement with the prediction that miR-206* can downregulate protein expression from the c.*22C>A allele, application of miR-206* reduced the pRL-4xA (mutant) signal to ∼59% of the signal obtained with pRL-4xC (wild-type). Similarly, miR-206* reduced the luminescence signal from the mutant construct to ∼57% of the signal obtained with non-specific control microRNA (Fig. 4B). Application of increasing concentrations of miR-206* (0, 10, 50 or 100 nm) showed a clear dose–response relationship between luminescence signals for pRL-4xA (mutant) and the microRNA (Supplementary Material, Fig. S3). Co-transfection of the pRL-4xA (mutant) construct with miR-206* and a miR-206* specific inhibitor abolished the effects of miR-206* (Fig. 4C). Experiments with luciferase vectors containing a single unit of the sequence around GFPT1 c.*22 (pRL-1xC (wild-type) and pRL-1xA (mutant)) yielded similar results. The pRL-1xA (mutant) signal was reduced to ∼66% of the signal obtained with pRL-1xC and miR-206* and to ∼61% of the signal obtained with pRL-1xA and control microRNA (Supplementary Material, Fig. S4).
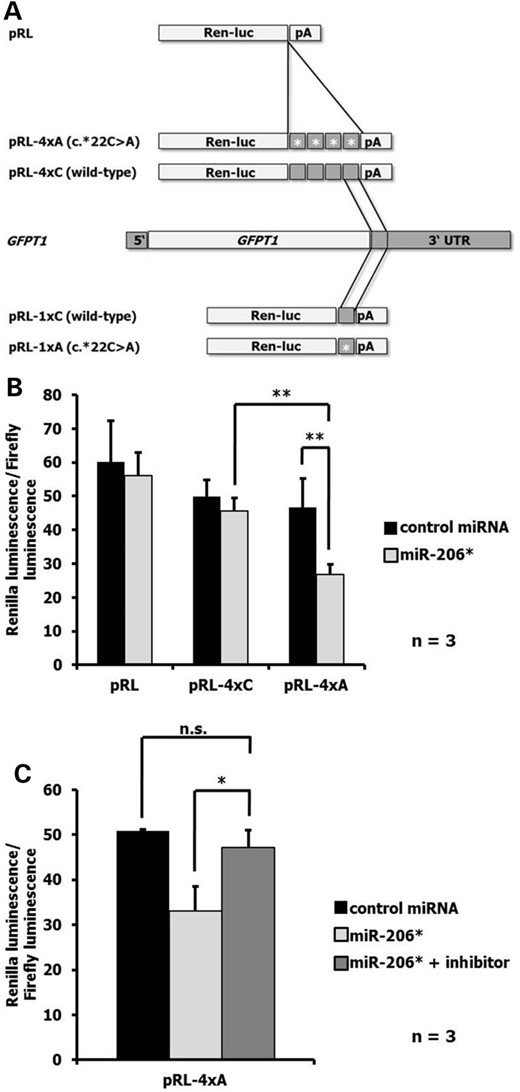
(A) Schematic representation of the Renilla luciferase (Ren-luc) expression vectors and the GFPT1 gene. (B) Renilla-to-firefly luminescence ratios observed when co-transfecting COS-7 cells with the indicated luciferase reporter (pRL, pRL-4xC (wild-type) and pRL-4xA (mutant) and either 100 nm control microRNA or miR-206* oligonucleotide. (C) Effect of an inhibitor against miR-206* on luminescence ratios obtained with pRL-4xA (mutant) and miR-206*. Data are derived from n = 3 independent experiments. P = 0.002, pRL-4xC + miR-206* versus pRL-4xA + miR-206*; P = 0.003, pRL-4xA + control miRNA versus pRL-4xA + miR-206*; P = 0.039, pRL-4xA + miR-206* versus pRL-4xA + miR-206* + inhibitor; n.s., not significant.
Restoration of GFPT1 expression upon miR-206* inhibition
Transfection of primary human myoblasts obtained from a patient with the c.22*C>A mutation (LGM9.3) with either αmiR-206* or non-specific control inhibitor (200 nm) revealed restoration of GFPT1 expression upon inhibition of miR-206*. In contrast, inhibition of miR-206* in myoblasts of a healthy control individual had no effect on the expression of GFPT1 (Fig. 5).
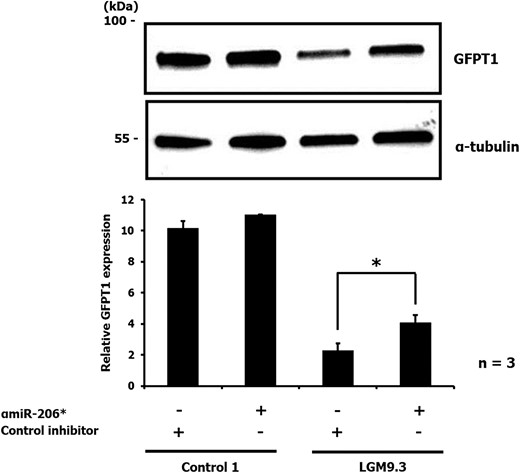
Immunoblot of GFPT1 in human primary myoblasts from a patient (LGM9.3: compound heterozygous for c.*22C>A and p.V199F) and a healthy donor (Control 1) transiently transfected with either 200 nm negative control or αmiR-206* inhibitor. An anti-α-tubulin antibody was used to ensure equal protein loading. Upper panel: representative result from three independent experiments. Lower panel: The intensities of the bands were measured and GFPT1 expression was normalized to the α-tubulin levels. P = 0.044, LGM9.3 + control inhibitor versus LGM9.3 + αmiR-206*.
Discussion
3′-UTRs play an important role in gene expression regulation by determination of mRNA stability/degradation, subcellular localization, nuclear export and translation efficiency (8–10). In this study we investigated the pathogenic relevance of a mutation in the 3′-UTR of the GFPT1 gene found in patients with CMS. We confirmed in silico predictions that this mutation can interfere with microRNA-mediated gene regulation.
Similar to other GFPT1 mutations, c.*22C>A has been shown to result in reduced protein expression but the underlying molecular mechanism had not yet been explored (3). We confirmed these observations by transfection experiments and expression studies involving patient-derived myoblast cultures. Because 3′-UTRs are frequent targets of microRNAs which are increasingly recognized as important regulators of gene expression (27), we used computer algorithms to predict microRNA binding sites in wild-type and mutated GFPT1 3′-UTR. We found a potential binding site for miR-206* that showed increased probability for microRNA binding when the mutation was present. Although there is no perfect miR-206*:mutated GFPT1 mRNA seed matching, the mutation strengthens a potential compensatory base pairing site in the 3′-region of the miR-206* (Fig. 3A). Even though perfect microRNA seed matches are often necessary for target regulation (28–31), recent studies have reported 3′-supplementary, 3′-compensatory pairing and ‘centered sites’ as new classes of microRNA target sites (32,33).
While it is known that miR-206 is highly expressed in human skeletal muscle and may play a critical role in myogenesis (24,34–37), there were no data on the expression of its star-counterpart miR-206*. We confirmed expression of miR-206* in skeletal muscle suggesting a physiologic role in this tissue (Fig. 3B). Moreover, overlapping expression profiles of miR-206* and GFPT1 also supported the assumption that the binding of this microRNA to the 3′-UTR of mutant GFPT1 mRNA might be pathogenetically relevant. In order to validate the interaction between the mutant GFPT1 transcript and miR-206* we made use of dual-luciferase reporter assays. When we co-transfected the mutant reporter constructs with miR-206* we observed a significantly diminished reporter signal compared with the signal obtained by co-transfection of the miR-206* and the wild-type reporter construct or by co-transfection of the mutant construct with control microRNA. Finally, a specific inhibitor for miR-206* blunted the effect of the microRNA on the luminescence signal related to the mutant construct (Fig. 4).
Our results support a model in which the point mutation c.*22C>A in the GFPT1 3′-UTR creates a target site for miR-206*, which influences GFPT1 expression. Mechanistically, we found that reduced GFPT1 protein levels resulted from repression of translation rather than degradation of the mRNA. Although most microRNAs act through destabilization of target mRNA, other microRNAs have been shown to affect translation. In a recent study about 16% of the analysed microRNAs decreased translational efficiency of the target mRNAs (38). So far, there are only a few reports which show a link between gene expression regulation through microRNA and hereditary human diseases. A mutation in the 3′-UTR of the HDAC6 gene was located in the seed region of mir-433 and interfered with post-transcriptional regulation in a patient with X-linked chondrodysplasia (39). In patients with Tourette syndrome, a variant in the 3′-UTR of the SLITRK1 gene was found to increase affinity to miR-189 leading to repression of SLITRK1 expression (40).
Recently, it has been suggested that mir-206 is upregulated following muscle injury and that increased miR-206 levels may slow progression of Duchenne muscular dystrophy (41). Moreover, miR-206 has been reported as a major regulator of repairing the neuromuscular junction following nerve injury (42). We observed upregulation of both miR-206 and miR-206* in GFPT1-diseased skeletal muscle. However, we also noted large interindividual differences of miR-206 and miR-206* levels among controls as well (Supplementary Material, Fig. S2). This might be related to different muscles used for surveying microRNA expression or—in case of affected patients—to different stages of the disease (24,43,44). Interfering with miR-206* activity could provide a therapeutic option for patients with the c.*22C>A mutation. Using reporter gene assays and patient-derived myoblasts we have provided evidence that miR-206* inhibitors efficiently blunt its effect on the GFPT1 c.*22C>A allele (Figures 4C and 5). In future studies, sponge constructs and/or antagomirs against miR-206* might be tested for recovering GFPT1 protein levels in skeletal muscle of patients with the GFPT1 c.*22C>A mutation.
In conclusion, our findings confirm that c.22*C>A is a causative mutation and provide further evidence that formation of illegitimate microRNA binding sites is a relevant pathomechanism. So far, only 0.2% of mutations causing Mendelian disorders have been estimated to reside within 3′-UTR (45). This is likely to be an underestimate and the number of phenotypes related to mutations in such so-far not well-recognized gene regions will soon increase thanks to next-generation sequencing studies. As to 3′-UTR regions, sequencing data will be ideally combined with new assays based on crosslinking immunoprecipitation (HITS-CLIP) used to identify functional microRNA targets (46).
Materials and Methods
Cell culture
C2C12 and COS-7 cells [American Type Culture Collection (ATCC, Manassas, VA, USA)] were cultured in Dulbecco's Modified Eagle's medium (DMEM; Gibco, Darmstadt, Germany) supplemented with 10% fetal calf serum (FCS), 2 mm glutamine and penicillin/ streptomycin (40 U/mL penicillin and 0.04 mg/mL streptomycin). Human myoblasts (Munich Tissue Culture Collection, Friedrich-Baur-Institute, Munich, Germany) were grown in skeletal muscle growth medium (SGM; PromoCell, Heidelberg, Germany) supplemented with SupplementalMix (Provitro, Berlin, Germany), 10% FCS, 1.5% 100 × Glutamax (Gibco) and 50 μg/ml gentamycin. For maturation into multinucleated myotubes, myoblasts were grown in SGM on culture dishes coated with laminin (Sigma-Aldrich, Steinheim, Germany) to about 90% confluence. Cells were induced to fuse and differentiate by replacing SGM with DMEM supplemented with 5% horse serum (fusion medium) for 7 days. All cell cultures were kept in a 37°C incubator with a humidified atmosphere of 5% CO2.
Determination of wild-type and mutant (c.*22C>A) GFPT1 mRNA levels by real-time quantitative reverse transcription PCR
Total RNA was isolated from snap-frozen muscle tissue or transiently transfected C2C12 cells using RNeasy kit (Qiagen, Hilden, Germany) according to the manufacturer's manual. The quality and quantity of total RNA was verified using a Nanodrop ND-1000 (PeqLab, Erlangen, Germany) spectrophotometer. Total mRNA (500 ng) was reverse-transcribed into cDNA by using iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA, USA) for mRNA isolated from muscle tissue and by using Omniscript Reverse Transcription Kit (Qiagen) for mRNA obtained from transfected C2C12 cells according to the manufacturer's instructions. Real-time quantitative reverse transcription PCR (real-time qRT-PCR) was performed in a 20 μl reaction volume with SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Each sample was amplified for 40 cycles in a 96-Well Optical Reaction Plate (Bio-Rad) using a CFX96 Real-Time System (Bio-Rad) and the cycle threshold (CT) was determined for each cDNA template. As a negative control, total RNA from each of the samples was run without reverse transcriptase (–RT). To correct for sample-to- sample variation, an endogenous control, Homo sapiens histone cluster 4 (hH4) or Mus musculus glyceraldehyde-3-phosphate dehydrogenase (mGAPDH), was amplified with the target. The expression levels of GFPT1 relative to those of hH4 or mGAPDH were calculated using the method (47). The primer pairs for mRNA expression were designed using published sequences (GFPT1: NM_002056.3; hH4: NM_175054.2 and mGAPDH: NM_008084.3). All oligonucleotide primers were synthesized at Metabion (Martinsried, Germany).
Plasmid constructs
The human GFPT1 coding region and 100 base pairs (bp) of the 3′-UTR were amplified from human skeletal muscle oligo(dT)-cDNA and inserted into the KpnI and NotI sites of the pEGFP-N1 plasmid (Clontech, Mountain View, CA, USA). To obtain GFPT1 without the green fluorescent protein (GFP) tag, the EGFP open reading frame was removed. The c.*22C>A mutant was generated by site-directed mutagenesis with mismatch primers (48). Correct orientation of the inserts and absence of PCR-induced mutations were verified by sequencing.
Transfection of myoblast cells
C2C12 cells were transfected with 3 μg of GFPT1-3′-UTR wild-type or mutant (c.*22C>A) constructs. Comparison of conserved sequences of miR-206* from Mus musculus (mmu) [MI0000249] and Homo sapiens (hsa) [MI0000490] revealed perfect sequence complementary between the seed regions and between the anchor regions (not shown). Therefore, similar effects in cells from human and mouse are expected in transfection experiments using plasmid constructs based on the human GFPT1 sequence. The cells were co-transfected with 0.3 μg pEGFP-N1 in order to use the expression of the GFP as a control for transfection efficiency. Lipofectamine 2000 (Invitrogen, Darmstadt, Germany) was used according to manufacturer's instructions. Primary human myoblasts were transfected with anti-miR-206* inhibitor (αmiRNA-206*) (200 nm; Riboxx, Radebeul, Germany) or Negative Control-N1 (200 nm; Riboxx) using GeneCellin transfection reagent (BioCellChallenge, Toulon, France) following manufacturer's recommendations. Cells were lysed 24 h after transfection and cell lysates were analysed by immunoblotting using antibodies against GFPT1, GFP and α-tubulin followed by appropriate secondary antibodies and chemiluminescence detection.
Preparation of cell lysates for protein analysis
Cultured cells were scraped off the plastic dish in phosphate buffered saline (PBS) on ice and pelleted by centrifugation (14,000 rpm, 5 min, 4°C). Subsequently, cell pellets were homogenized in lysis buffer (10 mm Tris–HCl, 1% sodium dodecyl sulfate (SDS), pH 7.4). Homogenized cell samples were then incubated at 95°C for 5 min. Debris was removed by 5 min centrifugation at 14,000 rpm and supernatants were used for immunoblot analysis.
Immunoblot analysis
Protein concentrations in the extracts were measured with the BCA protein assay (Thermo Scientific, Waltham, MA, USA). Protein samples were separated on 8% SDS-polyacrylamide gel electrophoresis gels. After electrophoresis, proteins were transferred to BioTrace™ NT Nitrocellulose Transfer Membrane (0.20 μm pore size; Pall Corporation, Crailsheim, Germany) for 1 h at 350 mA. Membranes were washed in Tris buffered saline with Tween20 (TBS-T) (Sigma-Aldrich) and blocked for 1 h at room temperature in TBS-T with 5% non-fat milk. Primary antibodies were incubated overnight at 4°C. The following antibodies were used: rabbit polyclonal anti-GFPT1 (Proteintech Group, Chicago, IL, USA; dilution 1:1,000), rabbit polyclonal anti-α-tubulin (Cell Signaling Technology, Leiden, The Netherlands; dilution 1:1,000) and rabbit polyclonal anti-GFP (Abcam, Cambridge, UK; dilution 1:10,000). Membranes were washed three times for 10 min with TBS-T and incubated with secondary goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP) (Invitrogen, dilution 1:5,000) or rabbit anti-goat IgG conjugated to HRP (Sigma-Aldrich, dilution 1:10,000) for 1 h at room temperature. Bands were detected with an enhanced chemiluminescence kit (GE Healthcare, Solingen, Germany).
In silico analysis
Bioinformatics tools on http://bioinfo.uni-plovdiv.bg/microinspector/ and http://www.mirbase.org/search.shtml were used to predict potential microRNA target sites in the 3′-UTR sequence of GFPT1.
Determination of miR-206 and miR-206* levels by real-time qRT-PCR
Small RNA (including microRNA) was isolated from snap-frozen muscle tissue, myoblasts and myotubes using miRNeasy Kit combined with the RNeasy MinElute Cleanup Kit according to the manufacturer's manual (Qiagen). MiR-206, miR-206* and miR-600 expression was measured by real-time qRT-PCR using the miScript-Reverse Transcription kit (Qiagen) and the miRNA-SYBR Green PCR kit (Qiagen). As a negative control, total RNA from each of the samples was run without reverse transcriptase. Relative microRNA expression levels were calculated using U6 snRNA (RNU6) as an internal control with the expression level in control myoblasts defined as 1. Evaluation of relative microRNA expression levels was performed based on the method (47).
Luciferase reporter assay
The pRL-TK (Promega, Madison, WI, USA) reporter vector containing a wild-type or mutant (c.*22C>A) 80 bp fragment of the GFPT1 3′-UTR was used for reporter assays in COS-7 cells. The partial 3′-UTR or multimers of it (Fig. 4A) were cloned into the XbaI restriction site downstream from the Renilla luciferase (RLuc) gene. The occurrence of secondary RNA structures that might interfere with microRNA binding in the final construct was excluded using RNAfold programme (49). An empty Firefly luciferase reporter vector (pGL4, Promega) was used as control. 0.8 × 105 cells per well in 24-well plates were transfected using Polyplus jetPEI transfection reagent (Biomol, Hamburg, Germany) following manufacturer's recommendations with a mixture comprising 200 ng of pRL-TK construct, 2 ng of pGL4 control vector and the mature microRNA mimic (100 nm; oan-miR-206*; Qiagen) or control microRNA (100 nm; Qiagen). As human miR-206* was not annotated, assays were performed using Ornithorhynchus anatinus (oan)-miR-206* after checking sequence homology using http://www.mirbase.org/. For blocking experiments 200 nm anti-miR-206* inhibitor (αmiRNA-206*) (Riboxx) were added. Luciferase expression was analysed 24 h after transfection using the dual-luciferase reporter assay system (Promega). The firefly reporter was measured first by adding Luciferase Assay Reagent II to generate a luminescent signal. After quantifying the luminescence, the reaction was quenched and the Renilla luciferase reaction was started by adding Stop & Glo Reagent. Luciferase expression was detected on a TriStar LB 941 reader (Berthold Technologies, Bad Wildbad, Germany). Relative reporter activity was obtained by normalization of Renilla luciferase activity to firefly luciferase activity. Each experimental condition was measured in triplicate and each assay was performed three times.
Statistical analysis
The data shown represent the mean ± standard deviation. Statistical significance was determined with two-tailed Student's t-test. A P-value of <0.05 was considered as statistically significant (*), P < 0.01 was considered as statistically very significant (**) and P < 0.001 was considered as statistically highly significant.
Supplementary Material
Supplementary material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (Ab 130/2-1) to A.A. and a grant from the Friedrich-Baur-Stiftung to J.S. J.S.M. and H.L. are supported by a grant from the Medical Research Council UK (reference G1002274, grant ID 98482). The Institute of Genetic Medicine in Newcastle is part of the MRC Centre for Neuromuscular Diseases. H.L. is supported by the European Union Seventh Framework Programme under grant agreement No. 305444 (RD-Connect) and 305121 (Neuromics). R.D. was supported by BMBF (FKZ 0312138A, FKZ 0316159), DFG (DA1296-2/1), European Social Fonds (FKZ V230-630-08-TFMV-F/S-035; ESF/IV-WM-B34-0011/08), (FKZ V630-F-075-2010/183; V630-S-075-2010/185; ESF/IV-WM-B34-0030/10), European Commission (FP7) SICA-HF (no. 241558), National Natural Science Fundation of China (no. 81370321) and Shanghai Pujiang Program (no. 13PJ1405800).
References
Author notes
Present address: Reference- and Translation Center for Cardiac Stem Cell Therapy (RTC), University Rostock, 18057 Rostock, Germany.
These authors have made equal contributions to the research.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}