




Abstract
ARX (the aristaless -related homeobox gene) is a transcription factor that participates in the development of GABAergic and cholinergic neurons in the forebrain. Many ARX mutations have been identified in X-linked lissencephaly and mental retardation with epilepsy, and thus ARX is considered to be a causal gene for the two syndromes although the neurobiological functions of each mutation remain unclear. We attempted to elucidate the causal relationships between individual ARX mutations and disease phenotypes by generating a series of mutant mice. We generated three types of mice with knocked-in ARX mutations associated with X-linked lissencephaly (P353R) and mental retardation [P353L and 333ins(GCG)7]. Mice with the P355R mutation (equivalent to the human 353 position) that died after birth were significantly different in Arx transcript/protein amounts, GABAergic and cholinergic neuronal development, brain morphology and lifespan from mice with P355L and 330ins(GCG)7 but considerably similar to Arx -deficient mice with truncated ARX mutation in lissencephaly. Mice with the 330ins(GCG)7 mutation showed severe seizures and impaired learning performance, whereas mice with the P355L mutation exhibited mild seizures and only slightly impaired learning performance. Both types of mutant mice exhibited the mutation-specific lesser presence of GABAergic and cholinergic neurons in the striatum, medial septum and ventral forebrain nuclei when compared with wild-type mice. Present findings that reveal a causal relationship between ARX mutations and the pleiotropic phenotype in mice, suggest that the ARX -related syndrome, including lissencephaly or mental retardation, is caused by only the concerned ARX mutations without the involvement of other genetic factors.
INTRODUCTION
The forebrain comprises a number of functionally and morphologically distinct types of neurons that, in coordination, regulate various aspects of cognition and behavior. ARX , thought to play an important role in forebrain development, is conserved in vertebrates and the ARX protein contains a prd -type homeodomain, four polyalanine tracts, an octapeptide and C-terminal ( aristaless ) domains ( 1 , 2 ). Located at Xp22.13, the ARX gene is considered to be a causative factor for X-linked lissencephaly and mental retardation ( 3–5 ), and more than 60 disease-related ARX mutations have been identified ( 6–8 ). These mutations are classified into two groups: one produces more severe lissencephaly and the other less severe mental retardation symptoms. The former group includes deletion, frameshift, nonsense and missense mutations within the homeobox and is associated with malformation syndromes such as lissencephaly, agenesis of the corpus callosum and ambiguous genitalia. The latter group includes elongation of the first or second polyalanine tract and missense mutations and is associated with syndromes without obvious malformations such as mental retardation, epilepsy and dystonia. The association of mental retardation with epilepsy is incidental since the patients sometimes had frequent and prolonged seizures, secondary neuronal injury and side effects of anticonvulsant. ARX mutations, however, provide a direct model for genetic association between epilepsy and mental retardation ( 9 ). Thus ARX is uniquely involved in a wide spectrum of neurodevelopmental disorders, ranging from mental retardation to lissencephaly.
Arx is expressed in the forebrain as well as in the testis, pancreas and skeletal muscles ( 3 , 10–12 ). It is involved in GABAergic neuron development in the cortex and striatum, in addition to cholinergic neuron development in the striatum, medial septum (MS) and ventral forebrain nuclei ( 3 , 13–15 ). Arx has both transcriptional activator and repressor domains ( 16 ) and functions as a transcriptional repressor ( 17 ). A portion of the genetic cascade of Arx in the forebrain has gradually been uncovered, with Dlx2 and Lmo1/Ebf3/Shox2 having recently been identified as upstream and target genes, respectively ( 18–20 ).
Important aspects to be addressed in the ARX -related syndromes, including lissencephaly and mental retardation, are (a) whether the single ARX mutations per se can cause one of the two syndromes and (b) whether there are specific causal relationships between individual ARX mutations and disease phenotypes. In the present report, we describe the generation of mice with three different knocked-in mutations associated with the ARX -related syndromes and show that the three mutations are causally involved in the syndromes and that the two mutations involved in the ARX -related mental retardation cause seizure and behavioral impairments through the abnormal development of GABAergic and cholinergic neurons.
RESULTS
Generation and general features of three ARX mutant mice
One of the mutations examined in this study was the mutation of a proline (353 in human ARX) in the conserved YPD (tyrosine–proline–aspartic acid) in the prd -type homeodomain of ARX. In humans, P353L develops X-linked myoclonic epilepsy with generalized spasticity and intellectual disability (OMIM300432), whereas P353R develops X-linked lissencephaly with ambiguous genitalia (XLAG, OMIM300215) ( 4 , 7 ). We also studied the addition of seven alanines to the first polyalanine tract, which leads to X-linked infantile spasms syndrome/West syndrome (OMIM308350) ( 4 ). We knocked-in the three mutations using homologous recombination; i.e. to study the first two mutations, we changed the equivalent proline (residue 355) of mouse Arx to either leucine or arginine, while seven GCG-triplets were inserted at residue 330 of the mouse Arx gene (Fig. 1 A and B). Using Southern analysis of genomic DNA from embryonic stem (ES) cells, we confirmed that each mutation was correctly introduced into the Arx gene (Fig. 1 C and D). Hemizygous mice ( Arxmt/Y ) obtained by crossing female heterozygous mice ( Arxmt/X ) with male wild-type mice ( ArxX/Y ) were used in the subsequent experiments [ Arxmt/Y mice: ArxP355R/Y (abbreviation: ArxPR/Y ), ArxP355L/Y ( ArxPL/Y ), Arx330ins(GCG)7/Y ( Arx(GCG)7/Y ) mice].
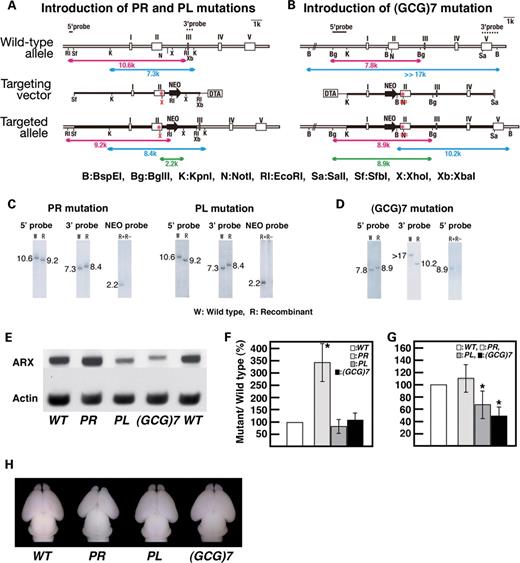
Generation and general features of ArxPR/Y , ArxPL/Y and Arx(GCG)7/Y mice. ( A and B ) Schematic representation of the wild-type Arx allele (top), targeting vector (middle) for introducing either the PL or PR mutations (A) and the (GCG)7 mutation (B), and the mutated allele after homologous recombination (bottom). The mutation points are indicated by (*). A new Xho I site is formed upon the introduction of the PL or PR mutations (A), whereas a Not I site is destroyed upon the introduction of the (GCG)7 mutation (B). ( C and D ) Southern blot analysis of the targeted ES clone with either the PL or PR mutations (C) and the (GCG)7 mutation (D). Genomic DNA from the ES cells was digested with either Eco RI for the PL/PR mutations (red bar in A) or Bgl II for the (GCG)7 mutation (red bar in B), and both digests were probed with a 5′-probe (C and D). Genomic DNA was then digested with Kpn I for the PL/PR mutations (blue bar in A) or Bam HI for the (GCG)7 mutation (blue bar in B), and both digests were probed with a 3′-probe (C and D). To confirm that each mutation was introduced, genomic DNA was either digested with Xho I (green line in A) and probed with a Neo-probe for the PL/PR mutations (C) or digested with Bgl II/ Not I (green bar in B) and probed with a 5′-probe for the (GCG)7 mutation (D). ( E ) Western blotting of Arx protein from the forebrains of three mutant embryos at E14. Arx protein from ArxPR/Y and ArxPL/Y embryos was the same length as the wild-type Arx, whereas the protein from Arx(GCG)7/Y embryos had a slightly higher molecular weight than that from the wild-type Arx. ( F ) Quantitative analysis of Arx transcript by real-time PCR. All values of the Arx transcript were normalized for Gapdh . The Arx transcript from ArxPR/Y embryos was approximately three times that of the wild-type Arx , whereas the Arx transcripts of ArxPL/Y and Arx(GCG)7/Y embryos were approximately the same as those of the wild-type Arx . ( G ) Semi-quantitative analysis of Arx protein by western blotting. All values of the Arx protein were normalized for actin. Arx protein levels from ArxPR/Y embryos were approximately the same as those from the wild-type Arx, whereas Arx protein levels in ArxPL/Y and Arx(GCG)7/Y embryos were 66.4 ± 22.6% ( P < 0.02, n = 5) and 48.4 ± 16.4% ( P < 0.001, n = 5) of those in the wild-type Arx, respectively. ( H ) Dorsal views of whole brains of newborn mice. ArxPR/Y mice had smaller brains and olfactory bulbs, whereas the neonatal brains of ArxPL/Y and Arx(GCG)7/Y mice were almost as large as those of ArxX/Y mice.
Arx protein from ArxPR/Y and ArxPL/Y embryos was the same length as that from the wild-type Arx, whereas the protein from Arx(GCG)7/Y embryos had a slightly higher molecular weight than that from the wild-type Arx owing to the addition of seven alanines (Fig. 1 E). We examined the expression amounts of the mutated Arx transcripts and Arx proteins using quantitative real-time PCR and semi-quantitative western blotting, respectively. The amount of Arx transcript from ArxPR/Y embryos was approximately three times that of the wild-type Arx (this was also confirmed by the endpoint PCR, unpublished data), whereas those from ArxPL/Y and Arx(GCG)7/Y embryos were approximately equal to that of the wild-type Arx transcript (Fig. 1 F). The amount of Arx protein in the ArxPR/Y embryo was approximately the same as in the wild-type Arx, whereas the protein from the ArxPL/Y and Arx(GCG)7/Y embryos was 40–60% that of the wild-type Arx; lower levels of protein were also found in another GCG mutant mouse, Arx432-455dup/Y (Fig. 1 G, unpublished data). The low levels may be based on susceptibility to proteolysis of the mutated Arx, while detailed analysis of the mechanisms responsible for expression amounts of Arx transcript and protein in the ArxPR/Y embryo is required.
ArxPR/Y mice died within 1 day after birth, as did Arx deficient ( Arx-/Y ) mice ( 3 ), whereas ArxPL/Y mice lived for more than 6 months. Most of the Arx(GCG)7/Y mice died within 3 months, but some of them survived for 5–6 months. Neonatal ArxPR/Y mice had smaller brains and olfactory bulbs, which were also seen in Arx−/Y mice ( 3 ), whereas the neonatal brains of ArxPL/Y and Arx(GCG)7/Y mice were nearly as large as those of ArxX/Y mice (Fig. 1 H).
Disturbed development of GABAergic and cholinergic neurons in embryonic and neonatal ArxPR/Y , ArxPL/Y and Arx(GCG)7/Y mice
Arx deficiency results in the loss of most of the tangential migration, except for that along the subventricular zone ( 3 ). The migration of GABAergic progenitor cells was examined using mutant mice cross-bred with heterozygous glutamate decarboxylase (GAD) 67-GFP (Δneo) mice, (GAD67 GFP/+ mice) ( 21 , 22 ), which demonstrated that most of the GABAergic interneuron precursors expressed Arx (Fig. 2 A–D). The migration modes in ArxPR/Y and ArxPL/Y embryos were very different from one another (Fig. 2 B and C). Arx + GABAergic progenitor cells of ArxPR/Y embryos failed to initiate migration at E12.5 (Fig. 2 B) and began to migrate only along the subventricular zone of the cortex at E14.5 (inset of Fig. 2 B). The total number of Arx + cells was 28.5 ± 3.4% ( P < 0.001, n = 3) of that found in wild-type cells in the cortical plate at P0 (Fig. 2 F). The Arx + GABAergic progenitor cells of ArxPL/Y and Arx(GCG)7/Y embryos began to migrate at E12.5 (Fig. 2 C and D), finally reaching 92.8 ± 8.7% and 88.4 ± 11.2% ( P < 0.02, n = 3), respectively, of the number of wild-type cells found in the cortical plate at P0 (Fig. 2 G and H). Thus, the PR mutation associated with XLAG resulted in severe defects in cortical tangential migration from the medial ganglionic eminence (MGE), whereas the PL and (GCG)7 mutations associated with ARX -related mental retardation resulted in only slight impairments.
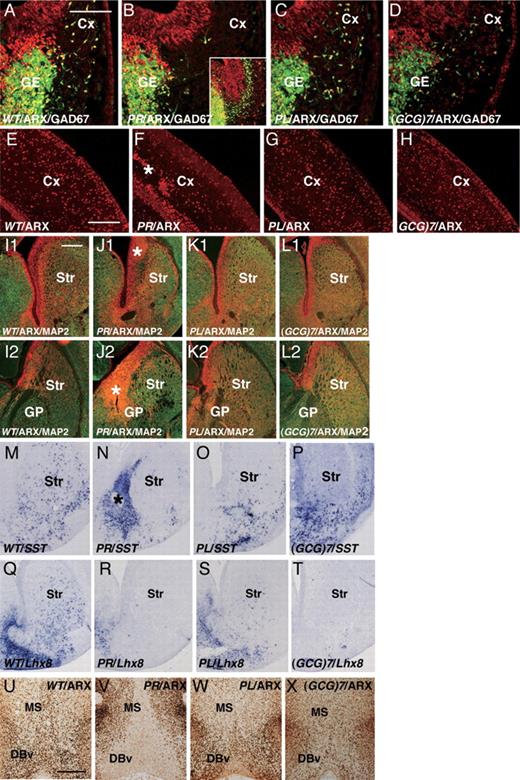
Characterization of the cortex (Cx), striatum (Str) and medial septum (MS) of embryonic and neonatal ArxPR/Y , ArxPL/Y and Arx(GCG)7/Y mice. ( A – D ) Cortical tangential migration in embryos (E12.5) crossbred with GAD67 GFP/+ mice. Arx + (red)/GAD67-GFP + (green) cells in the ArxX/Y embryos began to migrate from the ganglionic eminence (GE) (A), whereas no migration from the GE was seen in the ArxPR/Y embryos (B). Only late migration from the GE along the subventricular zone of the Cx was seen at E14.5 (inset of B). Arx + /GAD67-GFP + cells in ArxPL/Y and Arx(GCG)7/Y embryos migrated normally, although the number of cells was slightly reduced compared with that in the ArxX/Y embryos (C, D). ( E–H ) Arx + cells in the Cx at P0 (E–H). Significantly reduced numbers of Arx + cells were seen in the ArxPR/Y mice, and some of them were clustered at the subplate (F). Arx + cells were only slightly reduced in the ArxPL/Y and Arx(GCG)7/Y mice (G, H). ( I1 – L1 ) Anterior Str and ( I2 – L2 ) posterior Str, ARX (red) and MAP2 (green) expression in the Str at P0. The Arx + and MAP2 − ventricular zone of the anterior Str of ArxPR/Y mice showed increased thickness (* in J1), whereas no increase was seen in ArxPL/Y (K1) or Arx(GCG)7/Y mice (L1). The thickening of the ARX + and MAP2 + ventricular zone was also seen in the posterior Str of ArxPR/Y mice (* in J2), whereas no increase was seen in the ArxPL/Y (K2) and Arx(GCG)7/Y mice (L2). ( M – P ) SST + interneurons in the Str at P0. SST + interneurons were distributed throughout the Str of the ArxX/Y mice (M), whereas they gathered in the thickened ventricular zone of the striatum of ArxPR/Y mice (* in N) and were not present in the mantle zone of the Str (N). On the other hand, a small number of SST + interneurons were seen in the mantle zone of the Str of ArxPL/Y mice compared with ArxX/Y mice (O), whereas most of the SST + interneurons were seen in the ventricular zone of the Str in Arx(GCG)7/Y mice (P). ( Q–T ) Lhx8 expression in the Str at P0. Lhx8 expression was seen throughout the Str of ArxX/Y mice (Q), whereas no expression was seen in ArxPR/Y mice (R). Lhx8 expression was seen in the ventral half of the Str of ArxPL/Y mice (S), whereas no expression was seen in the Arx(GCG)7/Y mice (T). ( U – X ) Arx expression in the MS and vertical limbs of the nucleus of the diagonal band (DBv) at P0. ARX was expressed in the MS and DBv of the ArxX/Y mice (U), but no expression was seen in ArxPR/Y mice (V). Furthermore, significantly reduced Arx expression was observed in the ArxPL/Y and Arx(GCG)7/Y mice (W, X). MGE: medial ganglionic eminence, GP: globus pallidus. Scale bars: A–D, 100 µm; E–H, 250 µm; I1–T, 500 µm; U–X, 500 µm.
Arx deficiency results in a thickened subventricular zone of striatum and a loss of GABAergic interneurons in the striatum, suggesting the inhibition of both the radial migration of GABAergic projection neurons from the lateral ganglionic eminence and the tangential migration of GABAergic interneurons from the MGE ( 3 , 15 ). Arx + and MAP2 − ventricular and subventricular zones of the striatum were thicker in the ArxPR/Y mice at P0 (asterisk in Fig. 2 J1 and J2). Furthermore, somatostatin (SST) + cells, a subtype of GABAergic interneurons, accumulated in the subventricular zone of the ventral striatum (around the nucleus accumbens), but no SST + cells were detected in the mantle zone of the striatum (Fig. 2 N). Thus, both radial and tangential migrations were significantly suppressed in the ArxPR/Y mice, and a very similar situation was seen with the Arx−/Y mice ( 3 , 15 ). Radial migration in the ArxPL/Y and Arx(GCG)7/Y mice, on the other hand, was barely suppressed compared with that in the ArxPR/Y mice at P0 (Fig. 2 K1, K2, L1, L2), whereas tangential migration of striatal SST + cells was suppressed in a manner dependent on each mutation (Fig. 2 O and P). Thus, the PR mutation induced severe defects in striatum formation, which depends on both the radial and tangential migrations of presumptive GABAergic neurons, whereas the PL and (GCG)7 mutations caused impairments only in tangential migration of interneurons to the striatum.
Arx induces Gbx1 and Lhx8, making it essential for the development of cholinergic neurons; no cholinergic neurons were detected in the forebrain of the Arx−/Y mice ( 15 ). In ArxPR/Y and Arx(GCG)7/Y mice, no Lhx8 expression was found in the striatum at P0 (Fig. 2 R and T), and only a slight reduction in Lhx8 expression was detected in the striatum of ArxPL/Y mice (Fig. 2 S). ARX was expressed in the MS and the vertical limbs of the nucleus of the diagonal bands (DBv) of ArxX/Y mice at P0 (Fig. 2 U), whereas no expression was seen in ArxPR/Y mice (Fig. 2 V). Furthermore, the ArxPL/Y and Arx(GCG)7/Y mice showed much reduced ARX expression in the MS and DBv (Fig. 2 W and X). These Arx expression patterns in the MS and DBv of the three mutants were also seen for Lhx8 expression (data not shown).
Arx−/Y mice with a nonsense mutation that causes XLAG show a thinner cortical plate without a severely abnormal structure (such as inversion of cortical layers) at P0 ( 3 ). ArxPR/Y mice with the missense mutation that causes XLAG also exhibited a thinner cortical plate compared with ArxX/Y mice at P0 (85.2 ± 4.8% of wild-type, P < 0.01, n = 3. Fig. 3 A and B) The ratio of Tbr1 + deep layer ( 23 ) to whole cortical plate in ArxPR/Y mice was slightly higher than that of the wild-type (Fig. 3 G and H), whereas the Foxp1 + middle layer ( 24 ), located at the upper side of the cortical plate, exhibited no clear middle layer structure compared with wild-type (Fig. 3 E and F). Furthermore, the Satb2 + cells ( 25 ) were packed together in the uppermost layer, resulting in a thinner Satb2 + upper layer (Fig. 3 C and D). Thus, the abnormal cortical structure in ArxPR/Y mice at P0 suggests that fine regulation in both the proliferation of neuroepithelial cells in the ventricular zone ( 3 ) and the inside-out migration of post-mitotic neurons may be perturbed by Arx PR/Y protein expression in the embryonic ventricular/subventricular zones. Furthermore, this perturbation may be one reason why three cortical layers are formed in the human cortex with XLAG ( 26 , 27 ). No thin cortical plate or abnormal layer structure was observed in the ArxPL/Y and Arx(GCG)7/Y mice containing mutations that cause mental retardation (data not shown).
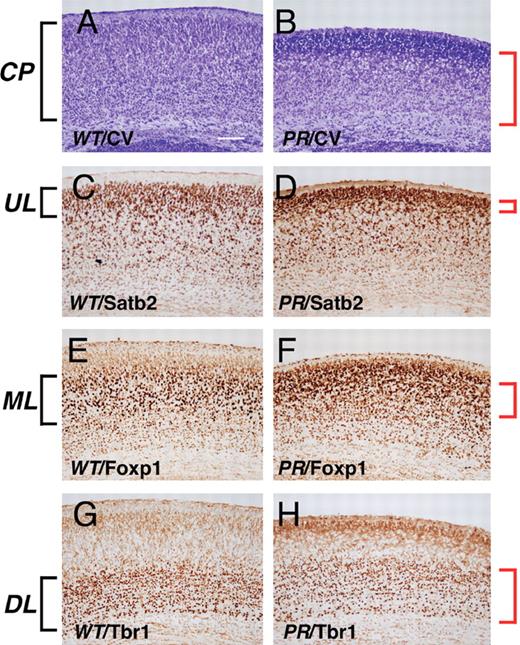
Aberrant cortical layer formation in ArxPR/Y mice. ( A and B ) Cresyl violet staining of the cortical plate of ArxX/Y (A) and ArxPR/Y (B) mice at P0. The thickness of the cortical plate was 85.2 ± 4.8% of wild-type ( P < 0.01, n = 3). ( C and D ) Satb2 imaging of the upper cortical layer of ArxX/Y (C) and ArxPR/Y (D) mice. Satb2 + cells in ArxPR/Y mice were packed together in the uppermost layer, resulting in a thinner Satb2 + upper layer than that of the wild-type. ( E and F ) Foxp1 imaging of the middle cortical layer of ArxX/Y (E) and ArxPR/Y (F) mice. The Foxp1 + middle layer of ArxPR/Y mice was located at the upper side of the cortical plate and exhibited no clear middle layer structure compared to wild-type. ( G and H ) Tbr1 imaging of the deep cortical layer of ArxX/Y (G) and ArxPR/Y (H) mice. The ratio of the Tbr1 + deep layer to the entire cortical plate in ArxPR/Y mice (60.3 ± 5.3%, P < 0.01, n = 3) was slightly higher than that of ArxX/Y mice (48.3 ± 3.8%, P < 0.01, n = 3). CP, cortical plate; UL, upper layer; ML, middle layer; DL, down layer. Scale bars: A–H, 100 µm.
The addition of seven alanines to the first polyalanine tract of Arx in vitro results in the formation of intranuclear inclusions and increased apoptosis ( 28 , 29 ), whereas the present in vivo study using Arx(GCG)7/Y embryos showed that no specific formation of intranuclear inclusions occurs in the migratory Arx + cells from ganglionic eminence at E12 and in the cortical Arx + cells at P0 ( Supplementary Material, Fig. S1A–D ). Furthermore, no increased apoptosis was detected in the ganglionic eminence at E12 ( Supplementary Material, Fig. S1E and F ). These observations were also seen in another mutation type in the GCG tracts, the duplication of polyalanine tract, ‘432–455dup’ (unpublished data). The absence of intranuclear inclusions in the present in vivo study could be based on differences in the in vivo and in vitro solubility/stability of Arx protein with extra alanine tracts.
In summary, the phenotype of mice with the PR mutation was entirely different from that of mice with the PL and (GCG)7 mutations; this strongly suggests that both groups of mutations can cause specifically developmental abnormalities in ARX -related lissencephaly (XLAG) or mental retardation. In order to further understand mutation effects on ARX -related mental retardation, we focused on the seizure, behavioral and neuronal analysis of the ArxPL/Y and Arx(GCG)7/Y mice at the post-natal stage.
Epilepsy in the ArxPL/Y and Arx(GCG)7/Y mice
Epilepsy is a common symptom in ARX -related mental retardation, and we examined the presentation of the epilepsy in both the types of mice. In the behavioral monitoring experiment, seizures were noted in 70% (7/10) of the Arx(GCG)7/Y mice at P1m (1-month-old mice), and one of them (1/10) died during status epilepticus. All seizures started with trembling of the limbs and progressed to tonic clonic convulsions, running fits and then complete loss of postural control and movement ( Supplementary Material, Movie ). Some of the mice exhibited hand-washing motions at the end of the seizures. Histologically, ectopically induced NPY expression in mossy fibers of the dentate gyrus was found in the brains of Arx(GCG)7/Y mice presenting seizures (Fig. 4 A1 and A2).
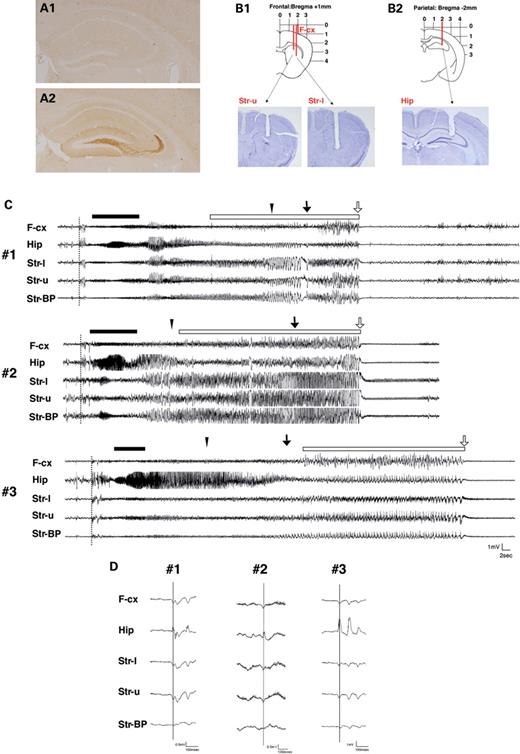
Presentation of seizures in Arx(GCG)7/Y mice. ( A1 and A2 ) Ectopic NPY expression in mossy fibers of the dentate gyrus. Before seizure (A1), after seizure (A2). ( B1 and B2 ) Diagrams of the electrode configuration superimposed on a coronal section of the brain. Local EEG was simultaneously recorded from the striatum (B1) and hippocampus (B2), together with the ipsilateral frontal EEG (B1). The recording sites were histologically verified in the cresyl violet staining sections. ( C ) Ictal EEG activity in three Arx(GCG)7/Y mice (#1, #2, #3). The seizure started (dotted line) simultaneously in the hippocampus, frontal cortex and striatum, followed by diffuse spike bursts. Next, 20–30 Hz spike bursts with waxing and waning appeared in the hippocampus (bar). Then there were continuous bursts of very high-voltage spikes (open bar) in the striatum, which abruptly changed into a long-lasting low-voltage activity with synchronous rhythmic theta waves. All seizures started with limb trembling that progressed to tonic clonic seizures (closed triangle), running fits (arrow) and complete loss of postural control and movement (open arrow). ( D ) Examples of EEG data at the beginning of a seizure in three Arx(GCG)7/Y mice (#1, #2, #3). The first spikes are simultaneously seen in the frontal cortex, hippocampus and striatum. F-cx, frontal cortex; Hip, hippocampus; Str-l, lower electrode in the striatum; Str-u, upper electrode in the striatum; Str-BP, bipolar in the striatum.
In three pairs of Arx(GCG)7/Y mice and their wild-type littermates, electroencephalographics (EEGs) were recorded in the frontal cortex, hippocampus and striatum together with the behavioral monitoring (Fig. 4 B1 and B2). In all three Arx(GCG)7/Y mice, the ictal EEG showed abnormal activities, although no spikes were found during the interictal period (Fig. 4 C). At the beginning of the seizure, EEG showed positive spikes in the hippocampus and negative spikes in the frontal cortex and striatum, followed by diffuse spike bursts (Fig. 4 D). The first spikes in 26 of 29 seizures were seen simultaneously in the hippocampus, frontal cortex and striatum. Then 20–30 Hz spike bursts with waxing and waning appeared in the hippocampus, and there were continuous bursts of very high-voltage spikes in the striatum. Finally, the bursts abruptly changed to long-lasting, low-voltage activity with synchronous rhythmic theta waves. Seizures often occurred in clusters, and the spikes were found between the seizure clusters.
In contrast, no ArxPL/Y mice died during the behavioral monitoring. Only one tonic seizure was seen at P1m in the ArxPL/Y mice (1/10), and the seizure was of shorter duration (30 s) than those seen in the Arx(GCG)7/Y mice (146 ± 57.9 s), but no seizures were observed in ArxPL/Y mice during EEG recordings. To determine the threshold of seizure and the relationship between age and seizure, the ArxPL/Y and ArxX/Y mice were challenged with bicuculline, a GABA A receptor inhibitor, at P1 m and P3–5m at a relatively low dosage (1.5 mg/kg). Myoclonic jerks were observed in most of the mice with both genotypes at both P1m and P3–5m, whereas generalized seizures developed less frequently in the ArxX/Y mice at P3–5m [Table 1 (A)]. Furthermore, significant differences were noted in the time from injection to onset of jerks and generalized seizures as well as in the number of deaths at both P1m and P3–5m [Table 1 (A) and (B)]. Thus, the seizure threshold was significantly lower in the ArxPL/Y mice than in the control ArxX/Y mice.
Seizure induction by bicuculline in ArxPL/Y mice
| Post-natal month old | Genotype | Mice with myoclonic jerks/total | Mice with generalized seizure/total | Deaths/total |
|---|---|---|---|---|
| (A) The number of mice displaying myoclonic jerks and generalized seizures and of deaths in the ArxPL/Y and ArxX/Y mice | ||||
| P1m | ArxP355L/Y | 6/6 | 6/6 | 6/6 |
| P3–5m | ArxX/Y | 6/6 | 4/6 | 1/6 |
| P | 1 | 0.4546 | 0.0152* | |
| P3–5m | ArxP355L/Y | 8/8 | 8/8 | 8/8 |
| ArxX/Y | 7/8 | 3/8 | 2/8 | |
| P | 1 | 0.0256* | 0.0070** | |
| (B) The onset of myoclonic jerks, generalized seizures and death from injection in the ArxPL/Y and ArxX/Y mice | ||||
| Post-natal month old | Genotype | Time myoclonic jerks start (min) | Time generalized seizure start (min) | Time of death (min) |
| P1m | ArxP355L/Y | 2.82 ± 0.60 | 3.58 ± 0.97 | 17.00 ± 9.69 |
| ArxX/Y | 5.43 ± 0.81 | 6.97 ± 1.83 | 18.00 ± 0 | |
| P | 0.0039** | 0.0105* | ||
| P3–5m | ArxP355L/Y | 3.34 ± 0.51 | 4.54 ± 1.62 | 9.00 ± 1.69 |
| ArxX/Y | 6.57 ± 4.16 | 9.53 ± 5.17 | 15.25 ± 1.06 | |
| P | 0.0038** | 0.0143* | ||
| Post-natal month old | Genotype | Mice with myoclonic jerks/total | Mice with generalized seizure/total | Deaths/total |
|---|---|---|---|---|
| (A) The number of mice displaying myoclonic jerks and generalized seizures and of deaths in the ArxPL/Y and ArxX/Y mice | ||||
| P1m | ArxP355L/Y | 6/6 | 6/6 | 6/6 |
| P3–5m | ArxX/Y | 6/6 | 4/6 | 1/6 |
| P | 1 | 0.4546 | 0.0152* | |
| P3–5m | ArxP355L/Y | 8/8 | 8/8 | 8/8 |
| ArxX/Y | 7/8 | 3/8 | 2/8 | |
| P | 1 | 0.0256* | 0.0070** | |
| (B) The onset of myoclonic jerks, generalized seizures and death from injection in the ArxPL/Y and ArxX/Y mice | ||||
| Post-natal month old | Genotype | Time myoclonic jerks start (min) | Time generalized seizure start (min) | Time of death (min) |
| P1m | ArxP355L/Y | 2.82 ± 0.60 | 3.58 ± 0.97 | 17.00 ± 9.69 |
| ArxX/Y | 5.43 ± 0.81 | 6.97 ± 1.83 | 18.00 ± 0 | |
| P | 0.0039** | 0.0105* | ||
| P3–5m | ArxP355L/Y | 3.34 ± 0.51 | 4.54 ± 1.62 | 9.00 ± 1.69 |
| ArxX/Y | 6.57 ± 4.16 | 9.53 ± 5.17 | 15.25 ± 1.06 | |
| P | 0.0038** | 0.0143* | ||
Values are mean ± SD.
* P < 0.05.
** P < 0.01.
Seizure induction by bicuculline in ArxPL/Y mice
| Post-natal month old | Genotype | Mice with myoclonic jerks/total | Mice with generalized seizure/total | Deaths/total |
|---|---|---|---|---|
| (A) The number of mice displaying myoclonic jerks and generalized seizures and of deaths in the ArxPL/Y and ArxX/Y mice | ||||
| P1m | ArxP355L/Y | 6/6 | 6/6 | 6/6 |
| P3–5m | ArxX/Y | 6/6 | 4/6 | 1/6 |
| P | 1 | 0.4546 | 0.0152* | |
| P3–5m | ArxP355L/Y | 8/8 | 8/8 | 8/8 |
| ArxX/Y | 7/8 | 3/8 | 2/8 | |
| P | 1 | 0.0256* | 0.0070** | |
| (B) The onset of myoclonic jerks, generalized seizures and death from injection in the ArxPL/Y and ArxX/Y mice | ||||
| Post-natal month old | Genotype | Time myoclonic jerks start (min) | Time generalized seizure start (min) | Time of death (min) |
| P1m | ArxP355L/Y | 2.82 ± 0.60 | 3.58 ± 0.97 | 17.00 ± 9.69 |
| ArxX/Y | 5.43 ± 0.81 | 6.97 ± 1.83 | 18.00 ± 0 | |
| P | 0.0039** | 0.0105* | ||
| P3–5m | ArxP355L/Y | 3.34 ± 0.51 | 4.54 ± 1.62 | 9.00 ± 1.69 |
| ArxX/Y | 6.57 ± 4.16 | 9.53 ± 5.17 | 15.25 ± 1.06 | |
| P | 0.0038** | 0.0143* | ||
| Post-natal month old | Genotype | Mice with myoclonic jerks/total | Mice with generalized seizure/total | Deaths/total |
|---|---|---|---|---|
| (A) The number of mice displaying myoclonic jerks and generalized seizures and of deaths in the ArxPL/Y and ArxX/Y mice | ||||
| P1m | ArxP355L/Y | 6/6 | 6/6 | 6/6 |
| P3–5m | ArxX/Y | 6/6 | 4/6 | 1/6 |
| P | 1 | 0.4546 | 0.0152* | |
| P3–5m | ArxP355L/Y | 8/8 | 8/8 | 8/8 |
| ArxX/Y | 7/8 | 3/8 | 2/8 | |
| P | 1 | 0.0256* | 0.0070** | |
| (B) The onset of myoclonic jerks, generalized seizures and death from injection in the ArxPL/Y and ArxX/Y mice | ||||
| Post-natal month old | Genotype | Time myoclonic jerks start (min) | Time generalized seizure start (min) | Time of death (min) |
| P1m | ArxP355L/Y | 2.82 ± 0.60 | 3.58 ± 0.97 | 17.00 ± 9.69 |
| ArxX/Y | 5.43 ± 0.81 | 6.97 ± 1.83 | 18.00 ± 0 | |
| P | 0.0039** | 0.0105* | ||
| P3–5m | ArxP355L/Y | 3.34 ± 0.51 | 4.54 ± 1.62 | 9.00 ± 1.69 |
| ArxX/Y | 6.57 ± 4.16 | 9.53 ± 5.17 | 15.25 ± 1.06 | |
| P | 0.0038** | 0.0143* | ||
Values are mean ± SD.
* P < 0.05.
** P < 0.01.
Impaired learning performance in both the ArxPL/Y and Arx(GCG)7/Y mice
Learning and memory of both types of mice were tested using a step-through passive avoidance task and two tasks on the eight-arm radial maze. A step-through passive avoidance task determines whether mice can retain an aversive memory of electric shocks. Naïve ArxPL/Y and Arx(GCG)7/Y mice showed no significant difference in latency to enter a darkened chamber compared with ArxX/Y mice ( P = 0.098 and P = 0.792, respectively, Mann–Whitney's U -test), which indicated that mobility and motivation for entry were not affected (Fig. 5 A and B). After the mice had experienced shocks, latency for entry was significantly shorter in both ArxPL/Y and Arx(GCG)7/Y mice than in ArxX/Y mice ( P = 0.021 and P < 0.001, respectively, Mann–Whitney's U -test), with a greater reduction of latency seen in Arx(GCG)7/Y mice (Fig. 5 A and B). These results indicated that neither type of mutant mouse could successfully acquire avoidance behavior through aversive experience.
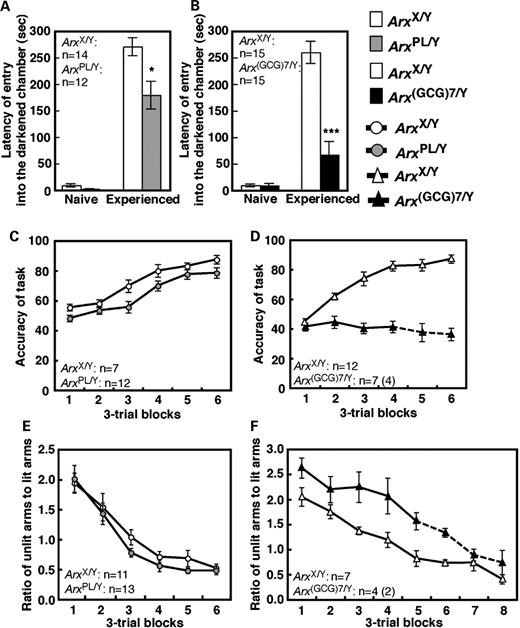
Defects in several aspects of learning and memory in ArxPL/Y and Arx(GCG)7/Y mice. ( A and B ) Latency to enter a darkened chamber in the passive avoidance task. The left set of bars represents results before the mice received electric shocks in the darkened chamber; the right set shows the results after shocks. A significant difference from the ArxX/Y mice is represented by * for P < 0.05 and *** for P < 0.001. ( C and D ) Accuracy of the win-shift task on the eight-arm radial maze, which we present as the percentage of chosen arms that were baited. The value is given as the mean of three trials. ( E and F ) The ratio of the unlit arm choices to lit arm choices in the win-stay task on the eight-arm radial maze. The value is given in the same manner as (C) and (D). The Arx(GCG)7/Y mice and ArxX/Y mice were statistically analyzed from blocks 1 to 4 in (D) and from block 1 to 5 in (F), using the number of the subjects shown in the figures. Mean values are shown on the solid line. The mean values on the broken line were calculated from the number of surviving Arx(GCG)7/Y mice in each block because some of them died during training. The number of subjects surviving throughout the tasks is shown in the parentheses.
The win-shift task on the eight-arm radial maze is a well-known hippocampus-dependent spatial learning task. To efficiently obtain all eight pellets at the end of each arm, mice must retain the spatial locations of pellet-retrieved arms within a trial. Performance was evaluated for accuracy, measured as the percentage of chosen arms that were baited. ArxPL/Y mice showed significantly improved accuracy with training, but their accuracy was slightly lower than that of ArxX/Y mice (genotype effect, F [1,17] = 9.12, P < 0.01; training effect, F [5,85] = 33.1, P < 0.0001; genotype × training interaction, F [5,85] = 0.702, P = 0.624) (Fig. 5 C). In Arx(GCG)7/Y mice, the accuracy was significantly poorer than in ArxX/Y mice (genotype effect, F [1,17] = 84.3, P < 0.0001; genotype × training interaction, F [3,51] = 10.8, P < 0.0001), as the Arx(GCG)7/Y mice showed no significant improvement in accuracy with training ( F [3,18] = 0.302, P = 0.824, one-way RMANOVA) (Fig. 5 D). These results indicate that both mutants showed inaccurate performance in pellet retrieval, with the Arx(GCG)7/Y mice showing more severe impairment than the ArxPL/Y mice. Because neither mutant used a non-spatial egocentric strategy by visiting each of the eight arms in turn ( Supplementary Material, Fig. S2 ), disabled spatial learning was thought to underlie the inaccurate performance of each type.
The win-stay task of the eight-arm radial maze is designed to evaluate striatum-dependent procedural learning in rodents. Mice can obtain a reward whenever they approach a lit arm. The arrangement of four lit arms is changed for every trial to avoid interfering with spatial memory. After acquiring a manner of approaching lit arms, mice approach the unlit arms less frequently. ArxPL/Y mice showed no significant difference in the ratio of unlit arm choices to lit arm choices compared with the ArxX/Y mice (genotype effect, F [1,22] = 1.78, P = 0.20; genotype × training interaction, F [7,154] = 0.43, P = 0.88) (Fig. 5 E). In contrast, Arx(GCG)7/Y mice showed a significantly higher unlit-to-lit ratio throughout training than the ArxX/Y mice did, although they did show a significantly decreased ratio with training (genotype effect, F [1,9] = 31.2, P < 0.001; training effect, F [4,36] = 13.4, P < 0.001; genotype × training interaction, F [4,36] = 0.19, P = 0.94) (Fig. 5 F). Thus, we concluded that the Arx(GCG)7/Y mice failed to acquire an association between the stimulus of light and the response of approaching, indicating a deficit in procedural learning.
In addition to several kinds of learning disabilities, we confirmed the impaired motor coordination in ArxPL and Arx(GCG)7/Y mice and the increased locomotor activity and anxiety-like behavior in Arx(GCG)7/Y mice ( Supplementary Material, Figs S3 and S4 ). Increased locomotor activity and anxiety-like behavior are thought to affect learning indices; however, the above-mentioned results in learning tasks were statistically independent from the increased locomotor activity and anxiety-like behavior in Arx(GCG)7/Y mice ( Supplementary Material, Table S1 ).
Slight and severe reduction of GABAergic neurons in the cortex and striatum, respectively, of ArxPL/Y and Arx(GCG)7/Y mice at P1m
We examined the post-natal distribution of GABAergic and cholinergic neurons based on the occurrence of seizure and impaired learning performance. In addition to being expressed during the embryonic period, Arx is expressed in each subtype of GABAergic interneurons, such as the parvalbumin (PV) + , SST + and neuropeptide Y (NPY) + cells and choline acetyltransferase (ChAT) + cholinergic neurons in the post-natal period ( Supplementary Material, Fig. S5 ). Furthermore, the findings at P1m described in this and the following sections were also confirmed at P2m (data not shown); thus, they were not due merely to a developmental delay.
We examined the number of GAD67 + neurons and subtypes of GABAergic interneurons in the somatosensory cortex of ArxPL/Y and Arx(GCG)7/Y mice at P1m. GAD67 + , SST + , NPY + and PV + interneurons exhibited no severe reduction in the cortex of ArxPL/Y and Arx(GCG)7/Y mice, with some exceptions, as suggested by the tangential migration of Arx + GABAergic neurons during the embryonic and neonatal stages ( Supplementary Material, Fig. S6 , Fig. 2 C, D, G, H). Tangential migration of striatal SST + cells was suppressed in a manner dependent on each mutation at P0 (Fig. 2 O and P). This observation suggested that these subtypes may be reduced at post-natal stages and there were clearly fewer SST + , NPY + and NOS (NO synthase) + interneurons in the striatum of the ArxPL/Y and Arx(GCG)7/Y mice (Fig. 6 A–I, M). On the other hand, there was only a slight reduction in the number of PV + interneurons in both mutants when compared with the other subtypes (Fig. 6 J–L, M). Furthermore, we found that SST + , NPY + and NOS + interneurons were localized to the ventral region and were rarely detected in the dorsal region of the striatum, whereas no regional differences were seen for PV + interneurons (Fig. 6 A–L). Thus, the number of SST + , NPY + and NOS + interneurons in the striatum of both mutants was significantly reduced when compared with that seen in the cortex, suggesting that striatal tangential migration was specifically suppressed in both mutants. We also found a subtype-specific reduction in the GABAergic interneurons in the basolateral amygdala of both mutants ( Supplementary Material, Fig. S7 ).
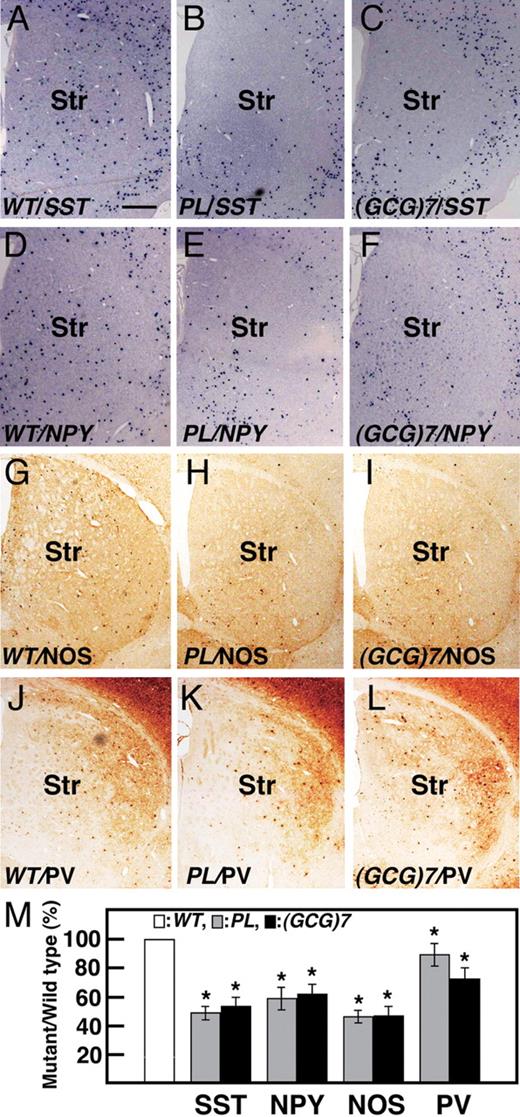
Reduction of neurons in each subtype of GABAergic interneurons in the striatum of ArxPL/Y and Arx(GCG)7/Y mice at P1m. SST + interneurons of ArxPL/Y (47.8 ± 4.8%, P < 0.001, n = 3. B, M ) and Arx(GCG)7/Y mice (52.4 ± 6.2%, P < 0.001, n = 3. C, M) were about half as many cells within the SST + interneurons as in ArxX/Y mice ( A ). The same tendency was seen for the NPY + ( D – F, M) and NOS + ( G – I, M) interneurons. In contrast, the reduction in PV + interneurons in ArxPL/Y (85.2 ± 4.2%, P < 0.01, n = 3. K, M) and Arx(GCG)7/Y (70.6 ± 7.3%, P < 0.002, n = 3. L, M) mice was small compared with the other subtypes. Values in (M) are normalized to the wild-type. Str: striatum. Scale bar: A–L, 500 µm.
Reduction of cholinergic neurons in the striatum, MS and ventral forebrain nuclei of ArxPL/Y and Arx(GCG)7/Y mice at P1m
Striatal ChAT + cholinergic interneurons were reduced by 50.1 ± 6.8% ( P < 0.001, n =3) in the ArxPL/Y mice, whereas no ChAT + interneurons were found in the Arx(GCG)7/Y mice, as expected from the Lhx8 expression at P0 (Figs 2 S, T and 7 B, C). Arx432-455dup/Y mice also had no ChAT + interneurons in the striatum (unpublished data). This observation suggests that migration and differentiation of cholinergic interneurons are fully suppressed by the elongation of the first and second polyalanine tracts and only partially by the PL mutation.
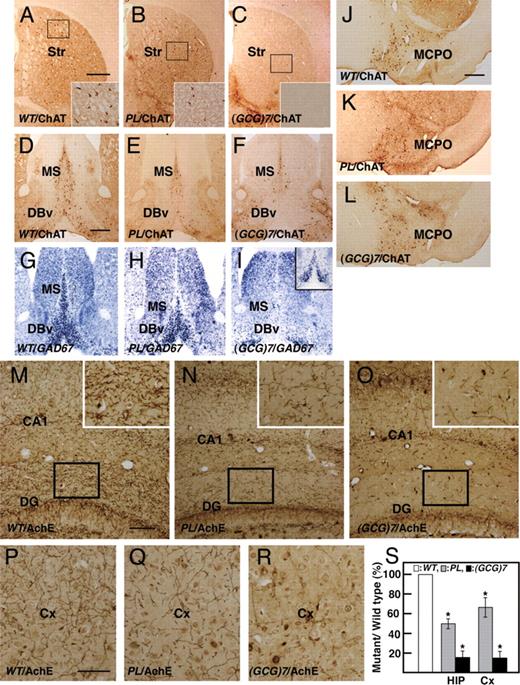
Reduction of cholinergic interneurons in the striatum and cholinergic and GABAergic projection neurons in the medial septum and ventral forebrain nuclei in ArxPL/Y and Arx(GCG)7/Y mice at P1m. ( A – C ) ChAT + cholinergic interneurons in the striatum. There were about half as many ChAT + interneurons in ArxPL/Y mice (B) as in ArxX/Y mice (A), whereas no cholinergic interneurons were found in Arx(GCG)7/Y mice (C). ( D–F ) ChAT + cholinergic projection neurons in the medial septum (MS) and vertical limbs of the nucleus of the diagonal bands (DBv). The number of neurons in the MS of ArxPL/Y mice was reduced to 53.2 ± 4.8% of that of ArxX/Y mice ( P < 0.001, n = 3, D, E), whereas it was reduced to 23.4 ± 4.0% of that of ArxX/Y mice in Arx(GCG)7/Y mice ( P < 0.001, n = 3, F). The number of neurons in the DBv of ArxPL/Y mice was reduced compared with the MS. ( G – I ) GAD67 + projection neurons in the MS and DBv. The number of neurons was severely reduced in the MS and DBv of ArxPL/Y mice (H), and no neurons were found in the MS or DBv of Arx(GCG)7/Y mice (I), except for GAD67 signals in the most anterior medial MS and DBv (inset of I). ( J – L ) ChAT + cholinergic projection neurons in the magnocelular preotic nucleus. The numbers of cholinergic projection neurons in ArxPL/Y( K, P) and Arx(GCG)7/Y (L, P) mice were 80.1 ± 8.5% ( P < 0.01, n = 3) and 68.4 ± 7.5% ( P < 0.001, n = 3), respectively, of that in ArxX/Y mice (J). ( M–S ) Acetylcholinesterase (AchE) + axonal arbors of cholinergic projection neurons in the hippocampus (M–O, S) and cortex (P–S). The total length of AchE + fibers per arbitral area in the hippocampus (molecular layer of dentate gyrus) of ArxPL/Y (N) and Arx(GCG)7/Y (O) mice was 50.5 ± 5.2% ( P < 0.01, n = 3) and 16.4 ± 6.7% ( P < 0.001, n = 3), respectively, of that in ArxX/Y mice (M). The total length of AchE + fibers per arbitral area in the somatosensory cortex of ArxPL/Y (Q) and Arx(GCG)7/Y (R) mice was 65.5 ± 9.8% ( P < 0.01, n = 3) and 15.8 ± 6.6% ( P < 0.001, n = 3), respectively, of that in ArxX/Y mice (P). Values in (S) are normalized to wild-type. Str: striatum, MS: medial septum, DBv: vertical limbs of the nucleus of the diagonal band, MCPO: magnocellular preoptic nucleus, DG: dentate gyrus, Cx: cortex. Scale bars: A–C, 500 µm; D–I, 500 µm; J–L, 500 µm; M–O, 100 µm; P–R, 200 µm; M–O, 100 µm; P–R, 50 µm.
Forebrain cholinergic projection neurons are found in the MS (Ch1), DBv and DBh (Ch2 and Ch3) and the basal magnocellular complex (Ch4), which is composed of the magnocellular preoptic nucleus (MCPO), substantia innominata, ventral pallidum and basal nucleus (NB). Ch1 and Ch2 provide major innervation to the hippocampus; Ch3 innervates the olfactory bulb and Ch4 innervates the cortex. ChAT + projection neurons were significantly reduced, to 53.2 ± 4.8% of the wild-type level in the MS of the ArxPL/Y mice ( P < 0.001, n = 3, Fig. 7 E), and were even more severely reduced in the Arx(GCG)7/Y mice (23.8 ± 4.0%, P < 0.001, n = 3, Fig. 7 F). Furthermore, the ChAT + neurons of ArxPL/Y and Arx(GCG)7/Y mice were reduced more severely in the DBv than in the MS (Fig. 7 E, F). GAD67 + neurons, another component of both the MS and DBv, were reduced in the ArxPL/Y mice and were not found in the Arx(GCG)7/Y mice, except for some expression in the most anterior MS and DBv (Fig. 7 H, I and inset of I). The number of ChAT + neurons in the MCPO of both mutants was decreased to ∼70% of the wild-type level (80.1 ± 8.5%, P < 0.01, n = 3 for ArxPL/Y mice, 68.4 ± 7.5%, P < 0.001, n = 3 for Arx(GCG)7/Y mice, Fig. 7 J–L), and the number of neurons in the NB of both mutants was also reduced (data not shown). Furthermore, we detected acetylcholinesterase (AchE) + axonal arbors of cholinergic projection neurons in the hippocampus and cortex, which are innervated by cholinergic projection neurons in the MS and basal magnocellular complex, respectively. A clear reduction in the AchE + axonal arbors was noted in the hippocampus and somatosensory cortex of ArxPL/Y mice (Fig. 7 N, Q, S) compared with ArxX/Y mice, in addition to a severe reduction seen in Arx(GCG)7/Y mice (Fig. 7 O, R, S). These data indicate aberrant formation of the Ch1/Ch2 (septohippocampal) and C4 systems in both mutant strains.
DISCUSSION
Diversity of ARX mutations and XLAG and mental retardation with epilepsy
One question to be addressed regarding the diversity of ARX mutations in the XLAG and mental retardation is whether the conditions are caused by individual mutations without involvement of any other genetic factors. We addressed this topic by introducing PR, PL and (GCG)7 mutations into the mouse genome. We found that phenotypes of ArxPR/Y mice are very similar to those of Arx−/Y mice with a truncated ARX mutation in XLAG ( 3 , 15 ). The abnormal cortical layer formation, abnormal structure of the striatum and deficiency of GABAergic neurons in the cortex and striatum caused by PR mutation closely mimic XLAG ( 5 , 26 , 27 ). Still, the mechanisms underlying the abnormal cortical formation specific to XLAG are not yet clear due to the perinatal death of both mouse models of XLAG ( 30 , 31 ). On the other hand, two structurally different mutations associated with ARX -related mental retardation, including the single amino acid replacement (PL) and elongation of polyalanine tract [(GCG)7], also caused a common phenotype of ARX -related mental retardation with epilepsy. Thus, the present study strongly suggests that PR and PL/(GCG)7 mutations independently cause lissencephaly and mental retardation, respectively, without the involvement of any other genetic factors.
The importance of the conserved YPD in the homeodomain was demonstrated in the ArxPR/Y and ArxPL/Y mutant mice ( 4 , 7 ). Proline in the YPD is predicted to have influence on homeodomain structure by providing the proper hydrophobic environment ( 32 ), whereas leucine and arginine have stronger and much weaker (hydrophilicity) hydrophobicity compared with proline, respectively ( 33 ). Thus, the hydrophobic environment suitable for the normal homeodomain structure and function may be destroyed by each mutation. It was surprising that the (GCG)7 mutation did not result in the specific formation of intranuclear inclusions and apoptosis that were observed in the in vitro functional test of the mutation ( 28 , 29 ) and this was also true of the Arx432–455dup/Y mice in our present study (unpublished data). The elongation of the polyalanine tract may function in the downregulation of ARX activity in cooperation with other functional domains of ARX such as homeodomain.
Seizure and ARX-positive neurons
Seizures occurred in most of the Arx(GCG)7/Y mice at P1m, and ∼10% died during an episode of status epilepticus, suggesting that the seizure caused the death. In contrast, only one ArxPL/Y mouse exhibited seizures, which were mild and occurred at 1 month of age. The observation that the longevity of ArxPL/Y mice was not different from that of ArxX/Y mice clearly shows that the seizure phenotype of the ArxPL/Y mice was milder than that of the Arx(GCG)7/Y mice. The onset latency and characteristics of the seizures in the ArxPL/Y mice following a low dose of bicuculline were significantly different from those of the ArxX/Y mice, suggesting that the ArxPL/Y mice had the epileptic phenotype. We conclude that both mutants show the epileptic phenotype that becomes apparent during late infancy to weaning age, albeit with different levels of severity in the two mutants. Interestingly, both types of mutants showed the epileptic phenotype with a severity of seizures that was similar to that in the corresponding human types. Most patients with the (GCG)7 mutations are diagnosed with infantile syndrome, whereas patients with the PL mutation develop myoclonic epilepsy ( 4 ). Epilepsy in patients with the (GCG)7 mutation is more severe than that in patients with the PL mutation.
Recently, it has been shown that specific abnormalities in the development or function of cortical GABAergic interneurons are linked to epilepsy ( 34 , 35 ). Arx−/Y mice exhibit interneuron migration deficiencies ( 3 ), and the ArxPR/Y mice in our present study were identical or very similar to Arx−/Y mice in this regard. Although Arx−/Y and ArxPR/Y mice did not exhibit seizures because they died around birth, the insufficient GABAergic interneuron migration may conceivably cause epilepsy in patients with lissencephaly due to the PR mutation. Surprisingly, we found that cortical and hippocampal GABAergic interneurons did not show a marked reduction in the ArxPL/Y or Arx(GCG)7/Y mice, but these mice did have seizures. Previous studies have shown that septal GABAergic projection neurons innervate GABAergic interneurons, and their cholinergic projection neurons form synapses with all neuron types in the hippocampus ( 36 , 37 ). It should also be noted that abolishing septal cholinergic projection neurons facilitated kindling in the hippocampus ( 38 ) and that inhibiting GABA A receptors expressed in GABAergic and cholinergic projection neurons in the septal region induced epileptiform activity in the hippocampus and seizures ( 39 ). Our results with the septum raise the possibility that seizures in ArxPL/Y and Arx(GCG)7/Y mice are caused by a loss of cholinergic and GABAergic projection neurons in that region.
We also revealed the loss of the GABAergic interneurons in the striatum. Much experimental evidence has suggested that the basal ganglia play an effective role in controlling seizures in animal epilepsy models ( 40 , 41 ), but the effects of the loss of striatal GABAergic interneurons in epilepsy remain enigmatic. We recorded EEGs in the striatum to examine its involvement in seizure generation and found that spike and burst patterns in the striatum were maintained at high voltage even after the voltage decayed in the hippocampus. Bipolar recording in the striatum also showed high-amplitude waves during the seizures, indicating local generation of epileptic activity. These EEG findings support a significant role of the striatum in seizure generation, although we could not detect the primary locus. The striatum receives input from large cortical areas. Epileptiform activity in the cortex evokes depolarizing bursts that are accompanied by action potentials in striatal projection neurons ( 42 ), and neocortical seizure-like EEG oscillations are evoked by tetanization of the striatum ( 43 ). Our results suggest that the loss of the GABAergic interneurons that inhibit projection neurons in the striatum may play a role in the epileptic network, inducing status epilepticus.
Impaired learning performance and ARX-positive neurons
Both ArxPL/Y and Arx(GCG)7/Y mice showed defects in several aspects of learning and memory. Passive avoidance is used as an experimental model of amnesia in rodents treated with cholinergic antagonists in the forebrain or striatum ( 44 ). Neither mutant could successfully acquire avoidance behavior, and the Arx(GCG)7/Y mice, with their severely impaired cholinergic projection neurons and interneurons, showed a more prominent defect than the ArxPL/Y mice. This observation suggests that these memory defects are due to cholinergic dysfunction in the forebrain of (GCG)7 and PL mutant mice. The win-shift task of the radial maze is a hippocampus-dependent spatial working or episodic-type memory task ( 45 ). Both types of mutant mice showed inaccurate performance on this task, with a more severe effect seen in the Arx(GCG)7/Y mice, which did not improve their performance with training. Significant impairment in win-shift performance was produced not by a single lesion in the cholinergic projection neurons in Ch1/2 but by a complex lesion in either the GABAergic projection neurons in Ch1/2 or the cholinergic projection neurons in Ch4 ( 46 ). Both types of mutant mice had a coincident reduction or loss of both projection neurons in Ch1/2 and cholinergic projection neurons in Ch4, which likely underlies their inaccurate performance. In the Arx(GCG)7/Y mice, this reduction or loss was more severe than in the ArxPL/Y mice. Moreover, a reduction in the hippocampal PV + interneurons could have affected win-shift performance ( 47 ) in the Arx(GCG)7/Y mice (data not shown, Supplementary Material, Fig. S6 ). Thus, these histological differences from the ArxPL/Y mice may have been manifested in the lack of improvement in win-shift performance of the Arx(GCG)7/Y mice. Win-stay performance, which represents striatum-dependent procedural learning ( 45 ), was impaired only in the Arx(GCG)7/Y mice in the present study. Cholinergic interneurons, which were reduced in both mutants, play an important role in procedural learning ( 48 , 49 ). The complete loss of the striatal cholinergic interneurons in Arx(GCG)7/Y mice likely led to their inferior win-stay performance. On the other hand, ArxPL/Y mice showed a less severe reduction in striatal cholinergic interneurons and unimpaired win-stay performance.
In conclusion, we reported a causal relationship between three Arx mutations and the pleiotropic phenotype using three generated mutant mice. The present mutant mice offer various research opportunities, including elucidation of the molecular properties of Arx cascade affected by each mutation and pathogenesis of aberrant neural systems caused by reduction of GABAergic and cholinergic neurons in the forebrain. Thus, these mutant mice could serve as excellent models for research on remarkable pleiotropy of XLAG and mental retardation with epilepsy and, moreover, for reversing the syndromes ( 50 ).
MATERIALS AND METHODS
The experiments were conducted in accordance with the animal care regulations of the Mitsubishi Kagaku Institute of Life Sciences, the National Institute of Neuroscience and the Tokyo Institute of Psychiatry.
Construction of the targeting vectors
We constructed three types of targeting vectors in which partial Arx fragments ( 3 ) containing the different mutations were linearly ligated to the Neo and DTA genes; these DNA fragments were then subcloned into pBluescript II (Fig. 1 A and B). The P355R mutation (CCT –> C g T) was introduced using the GENEEditor TM in vitro Site Directed Mutagenesis System (Promega, WI) and the sense primer 5′-CCAGCGGGAGGAA CTcGAG CGGGCTTTCCAGACGCACTACC- g TGACGTCTTCACCCAGG-3′ (the italicized bold lowercase letter shows the mutation site and the newly introduced Xho I site is in boldcase) (Fig. 1 A). The P355L (CCT –> C t T) mutation was introduced by PCR using the sense primer 5′-GGCTGCAGCGGCGGCGGCGGC-3′ and the antisense primer 5′-AAGACGTCA- a GGTAGTGCGTCTTCTGGAAAGCCCG CTCgAG TTCCTCCAGCTGGTAA-3′ (the italicized bold lowercase letter shows the mutation sit, and the newly introduced Xho I site is in boldcase) (Fig. 1 A). The addition of seven alanines into the first polyalanine domain was carried out by inserting 7 × (GCG) into a Not I site (325GCGGCCGC332), designated as the ‘330ins(GCG)7’ mutation (GCCGC –> GC ggcggcggcggcggcggcggc-g GC); this mutation was introduced by PCR using the sense primer 5′-GGCTGCAGC-GGCGGCG GCGGC ggcggcggcggcggcggcggcg GC AGCGGCGGCCACGGC-3′ and the antisense primer 5′-AAGACGTCAGGGTAGTGCGT-3′ (the italicized bold lowercase letters show the insertion sequence and the destroyed Not I site is in boldcase) (Fig. 1 B).
Transfection, screening and generation of three Arx mutant mice
To obtain the targeted ES cells, we used a positive–negative selection strategy in the CCE ES cells (a gift from Dr E. Robertson), which are derived from 129/Sv. We screened ES colonies by Southern blot analysis with a 5′ probe and a 3′ probe external to the genomic sequences contained in the targeting vector and a Neo gene (red and blue bars in Fig. 1 A and B). Furthermore, we confirmed the introduction of each mutation by Southern blot analysis using the introduced Xho I site in the PL and PR mutations or the lost Not I site in the (GCG)7 mutation (green bar in Fig. 1 A and B). We injected the positive ES clones into (C57BL/6J × BDF1) F1 or C57BL/6J blastocysts, which we then transferred into pseudopregnant female recipients. The resulting chimeric mice were bred with C57BL/6J females. Transmission of the targeted Arx locus was confirmed by Southern blot patterns. We obtained three Arx mutant mice ( Arxmt/Y : ArxPR/Y , ArxPL/Y , Arx(GCG)7/Y ), which were used in the subsequent analysis by crossbreeding with Arxmt/X and ArxX/Y mice. The Arxmt/Y hemizygotes derived from two independent targeted ES cell lines were phenotypically indistinguishable.
Genome DNA, Arx transcript and protein analysis
For Southern blots, we analyzed aliquots of genomic DNA from ES cells or tails of mice by 35 P-radioactive filter hybridization ( 3 ). For quantitative real-time PCR analysis, we carried out oligo-dT-primed reverse-transcription on aliquots of total RNA from the forebrain at E14.5 and used the single-strand cDNA products. Quantitative real-time PCR was performed with TaqMan primers on an ABI 7300 Real-Time PCR system (Applied Biosystems, CA, USA). All samples were analyzed in triplicate and were normalized relative to glyceraldehyde-3-phosphate dehydrogenase ( Gapdh ) expression, which served as an internal control for each sample. Gapdh expression was monitored using TaqMan rodent Gapdh control reagents (Applied Biosystems). We analyzed Arx expression using TaqMan Arx primers (Applied Biosystems). Expression levels were normalized to the wild-type for comparison. Data were analyzed with SDS 2.0 (Applied Biosystems) and Microsoft Excel, using the ΔΔCt method and the expression level of Gapdh as an internal reference for each sample. We carried out western blot analyses of RIPA buffer-extracted samples using an ARX polyclonal antibody ( 3 ). All samples were analyzed in triplicate and were normalized relative to actin expression, and the expression levels were normalized to the wild-type for comparison. Expression was detected by chemiluminescence (ECL, Amersham/GE Healthcare, NJ, USA), and the data were analyzed using the public domain Image J program developed by the US National Institutes of Health.
Histological analysis
Embryos were immersed and fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline for 12 h, and post-natal animals were fixed by cardiac PFA perfusion. Brains were post-fixed overnight, cryoprotected in a gradient of sucrose to 30%, frozen in embedding medium (OCT, Tissue-Tek) and serially sectioned at a thickness of 10 µm using a cryostat. For immunohistochemical staining, in situ hybridization and acetylcholinesterase histochemistry, four to six semi-sequential coronal sections per region (striatum, MS, MCPO, sensory cortex, hippocampus and basolateral amygdala) of three or four wild-type or mutant mice were selected according to the mouse brain atlas ( 51 ). Immunohistochemical staining and in situ hybridization were carried out as described ( 3 ). The antibodies used were anti-ARX antibody ( 3 ), anti-PV antibody (Sigma), anti-ChAT antibody (Chemicon/Millipore, MA, USA), anti-NOS antibody (Chemicon), anti-MAP2 antibody (Chemicon), anti-TBR1 antibody (abcam), anti-Foxp1 antibody (abcam) and anti-SATB2 antibody (Bio Matrix Research Inc.). Immunohistochemical signals were visualized by the following systems: (a) peroxidase-labeled secondary antibody/diaminobenzidine, (b) GFP-GAD67 and an anti-ARX antibody intensified with Alexa594-streptavidin (Molecular Probes/Invitrogen, CA, USA) and (c) an antibody intensified with FITC-labeled secondary antibody (Chemicon) and anti-ARX antibody intensified with Alexa594-streptavidin (Molecular Probe). The antisense probes were Lhx8 (a gift from Dr V. Pachnis), SST , NPY and GAD67 . Acetylcholinesterase histochemistry was performed as described previously ( 52 ). In each section, the number of positive cells per square micrometer in the observed region was calculated. Each value from the mutant mice was normalized to the wild-type mouse. Statistical analysis was done using a Student's t -test. Graphs show mean ± SEM.
Video monitoring of mice
Video monitoring was conducted for ArxPL/Y mice at the onset of weaning (P1m) and at the adult age (P5–6m) and for Arx(GCG)7/Y mice at P1m (each n = 10) in their home cages. The room light was on from 8:00 a.m. to 8:00 a.m., and their behavior was monitored with a CCD camera during the dark period (9–11 h/day) for 4 days under infrared light illumination. The data were stored in a hard disk recorder, and convulsive behavior was checked off-line by viewing the animals′ behavior on a screen.
Surface and depth EEG recording
ArxPL/Y and Arx(GCG)7/Y mice and their wild-type littermates ( n = 3 for each group) at P39–49 (P1m) were used for EEG recordings. Under chloral hydrate anesthesia (400 mg/kg, i.p.), four stainless wire electrodes (200 µm in diameter) were implanted using a stereotaxic apparatus (SR-8N, Narishige, Japan). One electrode was placed on the dura at the frontal cortex (F-cx; AP +1.0, L 2.0 from the Bregma; 51 ). Two wire electrodes were placed in the dorsal (Str-u; AP +1.0, L 2.0, DV 3.0) and ventral (Str-l; DV 4.0) parts of the striatum. Another wire electrode was inserted into the hippocampus (Hip; AP −2.0, L 2.0 DV 2.0) (Fig. 3 B1 and B2). The reference screw electrode was placed on the cerebellum surface. Lead wires from the electrodes were connected to a socket. And the electrodes and the socket were firmly attached to the skull with acrylic resin cement. After a recovery period of 3 days after surgery, an EEG was recorded for 1–2 days in a sound-attenuated experimental box. The data were amplified 1000 times (time constant; 0.1 s, low-pass filter; 100 Hz, AB-621G, Nihon Kohden, Japan) and monitored on a computer screen (Spike2 ver. 5, Cambridge Electronics Design, UK). The behavior of the mice was monitored with a CCD camera in the experimental box and recorded using a digital video camera (DCR-DVD301, Sony, Japan). The behavioral changes and accompanying EEG data were checked offline. After the experiments, electrode positions were verified histologically (Fig. 3 B).
Bicuculline treatment
To determine the seizure threshold and its change with age, ArxPL/Y and ArxX/Y mice at P1m (each n = 6) and P3–5m (each n = 8) received subcutaneous injections of 1.5 mg/kg bicuculline and their convulsive behavior was monitored for 30 min. The number of mice displaying myoclonic jerks and generalized seizures was recorded. In addition, the onset times of the first jerk and the first generalized seizure and the time of death from injection were recorded. Statistical differences in the number of mice and the time between injection and epileptic events were analyzed by Fisher's exact test and Mann–Whitney's U -test, respectively.
Experimental design for behavioral testing
The following behavioral tests were performed using ArxPL/Y , Arx(GCG)7/Y and ArxX/Y mice backcrossed to a C57BL/6J genetic background for F8–F9 generations: the wire-hanging test ( Supplementary Material, Fig. S3 ), the rotarod test ( Supplementary Material, Fig. S3 ), the open-field test ( Supplementary Material, Fig. S3 ), the step-through passive avoidance test (Fig. 4 ), the light–dark transition test ( Supplementary Material, Fig. S4 ) and two divided tasks on the eight-arm radial maze including the win-shift task and the win-stay task (Fig. 4 ). After weaning around post-natal week 4 (post-natal day 26–29), the mice were housed two to five animals to a cage in a room with a 12 h light/dark cycle (light beginning at 8:00 a.m.) with ad libitum access to food and water, except during the eight-arm radial maze tests. Behavioral testing began after three nights and was performed between 9:00 a.m. and 7:00 p.m. Almost all behavioral tests were carried out using identical mice. The Arx(GCG)7/Y mice had a short lifespan and were almost all deceased by 10 weeks of age. To avoid a decrease in the number of mice tested due to death, the two tasks on the radial maze were separated from the other tests and were begun as soon as possible after weaning.
Step-through passive avoidance
The step-through passive avoidance test was performed over 2 days using the testing apparatus (O'Hara & Co. Tokyo) as described by Karl et al . ( 53 ). On day 1, the mouse was placed into the illuminated chamber facing the door to the darkened chamber. The door then opened, and the latency of entry to the darkened chamber was measured. Electric shocks (0.2 mA) were delivered 3 s after the mouse entered. The mouse could freely escape from the darkened chamber with the door open and was allowed to explore these chambers for 60 s after the shocks. On day 2, the mouse was again placed into the illuminated chamber, and the latency of its entry to the darkened chamber was measured, with a 300 s cut-off time.
The win-shift task on the eight-arm radial maze
The win-shift task was performed in a manner similar to that described by Miyakawa et al . ( 54 ), using the eight-arm radial maze (O'Hara & Co.). The win-shift task was conducted during a habituation and training phase. Food deprivation (each mouse received only a 2.5–3.0 g pellet a day) began 3–4 days before the habituation phase. Over the 7 days of the habituation phase, the mouse underwent each of the three steps of pre-training in turn. In the first step, the mouse was allowed to freely explore the maze for 10 min without food reward. In the second and third steps, the protocol described by Miyakawa et al . ( 54 ) was followed. After the habituation phase, actual training began. All eight arms were baited with food pellets. The mouse was allowed to retrieve all eight pellets, with a 7 s confinement in the central platform before being presented with the next arm choice. The trial continued until the mouse had consumed all eight pellets or 10 min had elapsed. Training occurred once daily for 18 days. Image J RM software was used both to carry out the procedure and to analyze behavioral performance. Data for each mouse were averaged every three trials for statistical analysis.
The win-stay task on the eight-arm radial maze
The eight-arm radial maze was modified for the win-stay task. The eight arms had black opaque walls, and food dispensers were attached to the end of each arm to drop a sucrose pellet into the well. Light bulbs were set above the entrance to each arm facing the end of the arm. The apparatus was surrounded by a black curtain to restrict spatial cues and room light. The mice underwent the same food-deprivation schedule as in the win-shift task. The habituation and training phases were conducted for the win-stay task, during which four randomly selected arms were lit, but no more than three successive adjacent arms were lit on each trial. In the habituation phase, the mouse was allowed to freely explore the maze for 10 min a day for 3 days. The actual training was performed as described previously ( 55 ), following the habituation phase. A single pellet was placed in each well of the lit arms. The mouse was given two chances to retrieve the pellet in each lit arm and confined in the central platform for 7 s between each lit or unlit arm choice. This procedure was manually operated by remote control with video monitoring. One trial per day continued until the mouse had consumed all eight pellets or 10 min had elapsed. The latency to consume all pellets and the number of lit or unlit arms visited were measured for each trial. Data for each animal were averaged every three trials for statistical analysis.
Collection of behavioral indices for the eight-arm radial maze for the Arx(GCG)7/Y mice
Behavioral indices were collected, except for trials in which total choices were fewer than eight, which came from the total choices when a mouse retrieved all eight pellets without incorrect arm choices, because irregular and abnormal behavior occurred so often that the mouse had difficulty turning and choosing an arm. Additionally, if we saw an epileptic seizure, we allowed a 30–60 min interval before a trial to let the mouse calm down. Trials were sometimes stopped when it was apparent that a mouse could not move without difficulty.
Image analysis and statistical analysis
Software used for the behavioral tests (Image J OF, Image J LD, Image J EP and Image J RM) was modified on the public domain Image J program, developed by the US National Institutes of Health (available through O'Hara & Co). Each behavioral index was compared between the Arx mutant mice and ArxX/Y mice nursed by the heterozygous dams with the same kind of mutant allele. The data were analyzed using a two-tailed Student's t -test or two-way RMANOVA, unless noted otherwise, using Excel or GraphPad Prism 5. Graphs show mean ± SEM.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online .
FUNDING
This study was supported in part by the Research Grants (15B-4, 18A-5 and 21B-6) for Nervous and Mental Disorders from the Ministry of Health, Labour and Welfare (YG), and grants from the Organization for Pharmaceutical Safety and Research (MP-J) and the Institute of Biomedical Innovation (05-41), Japan (YG).
ACKNOWLEDGEMENTS
The authors express their appreciation to E. Robertson for providing the CCE ES cells and V. Pachnis for the Lhx8 ; K. Omichi, E. Iida, Y. Motegi, T. Hino, S. Kumagai and A. Takahashi for their technical support and K. Wada and K. Inoue for their encouragement.
Conflict of Interest statement . None declared.
REFERENCES
Author notes
The authors wish to be known that, in their opinion, they should be regarded to contribute equally to this work.
Present address: Department of Pediatrics, Graduate School of Medicine, University of Tokyo, Tokyo, Japan.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}