




Abstract
X-linked retinoschisis (XLRS) is a vitreo-retinal degeneration caused by mutations in the RS1 gene which encodes the protein retinoschisin (RS1), required for the structural and functional integrity of the retina. Data are presented from a group of 38 XLRS patients from Moorfields Eye Hospital (London, UK) who had one of 18 missense mutations in RS1. Patients were grouped based on mutation severity predicted by molecular modeling: mild (class I), moderate (intermediate) and severe (class II). Most patients had an electronegative scotopic bright flash electroretinogram (ERG) (reduced b/a-wave ratio) in keeping with predominant inner retinal dysfunction. An association between the type of structural RS1 alterations and the severity of b/a-wave reduction was found in all but the oldest group of patients, significant in patients aged 15–30 years. Severe RS1 missense changes were associated with a lower ERG b/a ratio than were mild changes, suggesting that the extent of inner retinal dysfunction is influenced by the effect of the mutations on protein structure. The majority of class I mutations showed no changes involving cysteine residues. Class II mutations caused severe perturbations due to the removal or insertion of cysteine residues or due to changes in the hydrophobic core. The ERG b/a ratio in intermediate cases was abnormal but showed significant variability, possibly related to the role of proline or arginine residues. We also conducted a second study, using a completely independent cohort, to indicate a genotype–ERG phenotype correlation.
INTRODUCTION
X-linked recessive retinoschisis (XLRS; OMIM312700) is a form of retinal dystrophy that affects males and causes splitting within the retinal layers, leading to early and progressive vision loss. Affected males typically have reduced b-wave amplitudes but relatively preserved a-waves on scotopic full-field electroretinogram (ERG) testing, indicating predominant inner retinal dysfunction (1). The retinal phenotype is associated with changes in the retinoschisin (RS) protein (2). RS1 is a retina-secreted, disulphide-linked, oligomeric protein which contains 224 amino acids. RS1 is expressed exclusively in the retina and in the pineal gland and functions as an adhesion molecule, preserving the structural and functional integrity of the retina.
Mutations in RS1 co-segregate with XLRS, providing strong evidence that the disease is caused by mutations in the RS1 gene (3). Approximately 170 unique sequence variations in the RS1 protein have been reported in XLRS patients to date (Leiden Open Variation Database; http://www.dmd.nl/rs/index.html). More than 60 mutations resulting in premature stop-codons and/or frameshifts are known to produce a truncated protein. Approximately 110 missense variants have so far been reported but the functional impact of most missense variants is not well known.
To date, most missense mutations have been found in exons 4–6 encoding the discoidin domain, establishing the significance of that part of the protein (1,2). Mature RS structure (23 kDa) consists of Rs1 (residues 24–63) and the highly conserved discoidin (residues 64–224) domains (4). The discoidin domain is present in single or multiple copies in extracellular and trans-membrane proteins implicated in cell–cell adhesion, and cell–cell interaction such as the coagulation factors V (FaV) and VIII (FaVIII), milk fat globule, neuropilin and neurexins, and is associated with galactose, collagen and phospholipid binding (5–7). Discoidin domains have a similar conserved β-barrel fold (8). In different species, they have 47 highly conserved residues (∼30% sequence identity), including two cysteines forming a disulphide bridge and several tryptophans maintaining a stable hydrophobic core of a discoidin domain (9).
Structural perturbations caused by missense mutations may have an impact on normal protein folding, solubility, stability, structure, function of protein associates or protein secretion. The structural effect of mutational changes can be analyzed in silico on the basis of three-dimensional structure, multiple alignments of homologous sequences and molecular dynamics (MD) (10,11). This approach suggests a potential for disease risk assessment at the atomic level (12). Recently, in 60 XLRS patients who share 27 missense mutations, the molecular models were correlated with retinal function as determined by the ERG a- and b-waves (13). The majority of RS1 mutations cause minimal structural perturbation and target the protein surface. The ERG b-wave/a-wave amplitude ratios in such cases were similar across younger and older subjects. Maximum structural perturbations from either the removal or insertion of cysteine residues or changes in the hydrophobic core were associated with greater difference in the b/a-ratio with age, with a significantly smaller ratio at younger ages. Such findings are analogous to the ERG changes with age observed in RS1-knockout mice that have no RS1 protein expression. The molecular modeling suggested an association between the predicted structural alteration and/or damage to RS and the severity of XLRS as measured by the ERG, analogous to the RS1-knockout mouse (14).
To test the validity of this approach, we have now compared the molecular models with the electrophysiological features in a cohort of 38 XLRS patients from Moorfields Eye Hospital (London, UK) having one of 18 RS1 missense variants (15).
RESULTS
Comparsion of measured and computed disease severity
The full-field ERG b/a-ratios in patients with missense changes ranged from 0.51 to 1.35 (normal 5th and 95th percentiles: 1.21–1.91) for scotopic (DA 11.0) and from 0.69 to 3.97 (normal 5th and 95th percentiles: 2.69–5.32) for photopic (LA 3.0) responses.
Patients were grouped based on mutation severity predicted by molecular modeling and the computed impact (CI) score (see Section ‘Methods’). The relationships between ERG b/a-wave ratios and the CI score for different missense mutations are shown in Fig. 1A and B. The mildest ERG abnormalities were associated with the lowest (class I) CI scores. The most severe ERG abnormalities were associated with the highest CI (class II) scores and were of similar severity to those associated with null mutations (Fig. 1C and D). Patients with missense variants of intermediate (moderate) CI had a wider range of ERG phenotypes than the other two groups (see Section ‘Discussion’) but tended to have b/a-ratios that were slightly closer in magnitude to those of the class I than class II groups (Fig. 1 A and B).
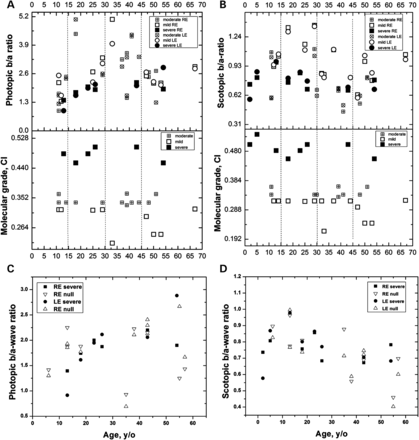
ERG b /a-wave amplitude ratios are shown for patients with XLRS as a function of age. Photopic (A) and scotopic (B) b/a-wave ratios and computed impact are shown in the top and bottom panels, respectively, as a function of age for 34 XLRS patients carrying 18 missense mutations, respectively. Squares and circles represent b/a-wave ratios for right (RE) and left (LE) eyes, respectively. Open, crossed and filled symbols correspond to computed mild, moderate and severe XLRS phenotypes, respectively. Vertical broken lines divide the age groups: <15, 15 to <30, 30 to <45, 45+ years. Groups according to the severity of missense mutations as evaluated using the computed impact (CI): mild (<0.32), moderate (0.32 to <0.4) and severe (0.4+). The photopic (C) and scotopic (D) b/a-wave amplitude ratios for the severe missense changes are plotted with the values for nonsense (null) mutations Table (2) as a function of age. Filled squares and circles show b/a-wave ratios for patients with severe missense changes measured in right (RE) and left (LE) eyes, respectively. Open triangles show b/a-wave ratios for patients with severe nonsense changes.
Severe nonsense changes in the RS1 protein
| Mutation | Protein | Age, years old | Scotopic b/a-ratio | Photopic b/a-ratio | ||
|---|---|---|---|---|---|---|
| RE | LE | RE | LE | |||
| Deletion 416delA in exon 5 | Truncation | 6 | 0.90 | 0.83 | 1.42 | 1.30 |
| Exon 1 deletion | Null | 13 | 0.96 | 1.00 | 2.25 | 1.93 |
| Substitution 103C > T | Truncation, p. Gln35X | 13 | 0.77 | 0.77 | 1.88 | 1.86 |
| Exon 1 deletion | Null | 18 | 0.81 | 0.74 | 1.88 | 1.77 |
| Splice site 184 + 2T > G | No protein | 25 | 0.72 | 0.74 | 1.71 | 1.70 |
| Insertion c.579insC | Truncation, p.His194fsX263 | 35 | 0.88 | 0.71 | 0.93 | 0.69 |
| Splice site IVS1 + 2T > C | No protein | 38 | 0.56 | 0.59 | 2.23 | 2.11 |
| Exon 1 deletion | Null | 43 | 0.69 | 0.72 | 2.10 | 2.41 |
| Exon 1 deletion | Null | 43 | 0.71 | 0.75 | 2.15 | 2.29 |
| Duplication 579dupC | Truncation | 49 | 0.88 | 0.83 | 2.54 | 1.74 |
| Deletion 416delA in exon 5 | Truncation | 55 | 0.46 | 0.40 | 1.25 | 2.67 |
| Exon 1 deletion | Null | 57 | 0.70 | 0.60 | 1.43 | 1.67 |
| Mutation | Protein | Age, years old | Scotopic b/a-ratio | Photopic b/a-ratio | ||
|---|---|---|---|---|---|---|
| RE | LE | RE | LE | |||
| Deletion 416delA in exon 5 | Truncation | 6 | 0.90 | 0.83 | 1.42 | 1.30 |
| Exon 1 deletion | Null | 13 | 0.96 | 1.00 | 2.25 | 1.93 |
| Substitution 103C > T | Truncation, p. Gln35X | 13 | 0.77 | 0.77 | 1.88 | 1.86 |
| Exon 1 deletion | Null | 18 | 0.81 | 0.74 | 1.88 | 1.77 |
| Splice site 184 + 2T > G | No protein | 25 | 0.72 | 0.74 | 1.71 | 1.70 |
| Insertion c.579insC | Truncation, p.His194fsX263 | 35 | 0.88 | 0.71 | 0.93 | 0.69 |
| Splice site IVS1 + 2T > C | No protein | 38 | 0.56 | 0.59 | 2.23 | 2.11 |
| Exon 1 deletion | Null | 43 | 0.69 | 0.72 | 2.10 | 2.41 |
| Exon 1 deletion | Null | 43 | 0.71 | 0.75 | 2.15 | 2.29 |
| Duplication 579dupC | Truncation | 49 | 0.88 | 0.83 | 2.54 | 1.74 |
| Deletion 416delA in exon 5 | Truncation | 55 | 0.46 | 0.40 | 1.25 | 2.67 |
| Exon 1 deletion | Null | 57 | 0.70 | 0.60 | 1.43 | 1.67 |
XLRS, X-linked retinoschisis; RS1, retinoschisin; ERG, electroretinogram; MD, molecular dynamics.
Severe nonsense changes in the RS1 protein
| Mutation | Protein | Age, years old | Scotopic b/a-ratio | Photopic b/a-ratio | ||
|---|---|---|---|---|---|---|
| RE | LE | RE | LE | |||
| Deletion 416delA in exon 5 | Truncation | 6 | 0.90 | 0.83 | 1.42 | 1.30 |
| Exon 1 deletion | Null | 13 | 0.96 | 1.00 | 2.25 | 1.93 |
| Substitution 103C > T | Truncation, p. Gln35X | 13 | 0.77 | 0.77 | 1.88 | 1.86 |
| Exon 1 deletion | Null | 18 | 0.81 | 0.74 | 1.88 | 1.77 |
| Splice site 184 + 2T > G | No protein | 25 | 0.72 | 0.74 | 1.71 | 1.70 |
| Insertion c.579insC | Truncation, p.His194fsX263 | 35 | 0.88 | 0.71 | 0.93 | 0.69 |
| Splice site IVS1 + 2T > C | No protein | 38 | 0.56 | 0.59 | 2.23 | 2.11 |
| Exon 1 deletion | Null | 43 | 0.69 | 0.72 | 2.10 | 2.41 |
| Exon 1 deletion | Null | 43 | 0.71 | 0.75 | 2.15 | 2.29 |
| Duplication 579dupC | Truncation | 49 | 0.88 | 0.83 | 2.54 | 1.74 |
| Deletion 416delA in exon 5 | Truncation | 55 | 0.46 | 0.40 | 1.25 | 2.67 |
| Exon 1 deletion | Null | 57 | 0.70 | 0.60 | 1.43 | 1.67 |
| Mutation | Protein | Age, years old | Scotopic b/a-ratio | Photopic b/a-ratio | ||
|---|---|---|---|---|---|---|
| RE | LE | RE | LE | |||
| Deletion 416delA in exon 5 | Truncation | 6 | 0.90 | 0.83 | 1.42 | 1.30 |
| Exon 1 deletion | Null | 13 | 0.96 | 1.00 | 2.25 | 1.93 |
| Substitution 103C > T | Truncation, p. Gln35X | 13 | 0.77 | 0.77 | 1.88 | 1.86 |
| Exon 1 deletion | Null | 18 | 0.81 | 0.74 | 1.88 | 1.77 |
| Splice site 184 + 2T > G | No protein | 25 | 0.72 | 0.74 | 1.71 | 1.70 |
| Insertion c.579insC | Truncation, p.His194fsX263 | 35 | 0.88 | 0.71 | 0.93 | 0.69 |
| Splice site IVS1 + 2T > C | No protein | 38 | 0.56 | 0.59 | 2.23 | 2.11 |
| Exon 1 deletion | Null | 43 | 0.69 | 0.72 | 2.10 | 2.41 |
| Exon 1 deletion | Null | 43 | 0.71 | 0.75 | 2.15 | 2.29 |
| Duplication 579dupC | Truncation | 49 | 0.88 | 0.83 | 2.54 | 1.74 |
| Deletion 416delA in exon 5 | Truncation | 55 | 0.46 | 0.40 | 1.25 | 2.67 |
| Exon 1 deletion | Null | 57 | 0.70 | 0.60 | 1.43 | 1.67 |
XLRS, X-linked retinoschisis; RS1, retinoschisin; ERG, electroretinogram; MD, molecular dynamics.
Figure 2 summarizes the ERG findings across different age ranges and CI groups under photopic and scotopic conditions. A statistically significant difference between class I (mild) and class II (severe) groups was found for both photopic (P-value = 0.002) and scotopic (P-value = 0.02) ERGs for those aged between 15 and 30 years (Fig. 2A and B; Supplementary Material, Tables S1 and S2 show corresponding P-values for multiple comparisons). In addition, the moderate group also showed a significant difference from the severe group for those aged 15–30 years. Regression analysis demonstrated that mean photopic b/a-wave ratios and mean photopic b-wave amplitudes were significantly lower for class II than for class I mutations in each of the three oldest age groups (P < 0.04 in all cases). In addition, the mean photopic b-wave peak times were significantly longer in those with class II mutations than in those with class I (mild) mutations in the two oldest age groups (P < 0.01). The ERG a-wave parameters showed no association with predicted severity of mutation. Mean scotopic b/a-wave ratios were significantly lower in individuals with class II compared with class I variants for one age group only (15 to < 30 years; P < 0.0001; Fig. 2). In addition, the mean scotopic a-wave amplitude, b-wave amplitude (ages 15 to <30), a-wave peak time (ages <15, 15 to <30) and b-wave peak time (ages <15, 15 to <30, 30 to <45) were significantly lower in those with severe than in those with mild mutations.
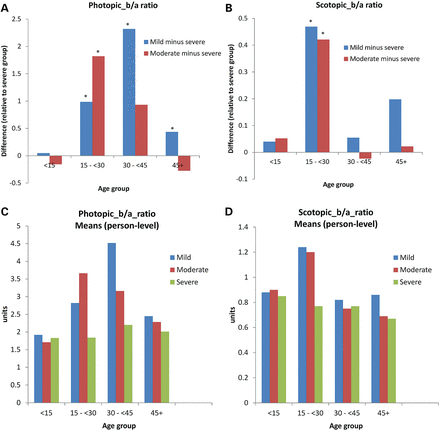
Relationships between mild (class 1) and severe (class II) missense mutations and ERG b/a ratios in older and younger XLRS patients. Photopic (A) and scotopic (B) responses were examined using differences for mild and moderate mutations relatively the severe group. A statistically significant difference between class I and class II groups exists for photopic (P-value = 0.002) and scotopic (P-value = 0.02) responses for the younger patients (15–30 years) (labeled by star*). A comparison of mean b/a-ratios for mild, moderate and severe mutations in four age groups is shown for photopic (C) and scotopic (D) responses. Groups in severity of missense mutations as evaluated using the computed impact (CI): mild (<0.32), moderate (0.32 to <0.4) and severe (0.4+).
Comparison of mild and moderate severity groups revealed no statistically significant differences except for photopic b/a ratio for age 30 to <45, which were 1.39 units higher in the mild than in the severe group (P = 0.002) (Supplementary Material, Fig. S1). Photopic responses showed no difference between the mild and moderate groups, with b/a ratios of 2.71 ± 1.05 versus 2.71 ± 1.11 (RE) and 2.58 ± 0.68 versus 2.71 ± 1.21 (LE), respectively. The b/a ratios were ∼30% lower in the severe group; 1.85 ± 0.27 (RE) and 1.92 ± 0.65 (LE). Corresponding changes for scotopic responses were smaller but showed the same trend.
Comparison with previously published data
The relationships between ERG b/a-wave ratios and classes of missense change were also examined in the two extreme groups (class I and class II mutations) according to age to allow comparison with an earlier study that used the same classification (Fig. 3, Supplementary Material, Table S3). A previously described method was used to minimize the effect of statistical errors and to improve a signal-to-noise ratio in experimental ERG data (13). The younger group was aged 23–33 years and the older group 34 years or over. The CI score values averaged within groups were similar for scotopic (class I = 0.31; class II = 0.39) and for photopic (class I = 0.32; class II = 0.38) responses. The b/a wave ratio associated with class II mutations was greater in the older than in the younger group, with the b-wave unchanged as the a-wave declined, similar to that observed in the Rs1b-KO mouse (14) and in the patient cohorts published previously (1,32). The a-wave amplitude decreased with increasing age for both class I and class II mutations, and the b-wave amplitude decreased with age for class I mutations but showed no relationship with age among those with class II mutations (Supplementary Material, Table S3).
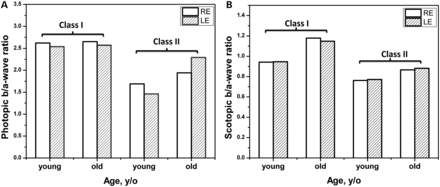
Scotopic and photopic ERG average b/a-wave ratios as a function of average patient age and severity of the XLRS phenotype. Average b/a-wave ratios obtained for patients divided into four groups by missense mutation classes (class I = mild, class II = severe) and by patient age (young and old) defined similarly to that shown in the Supplementary Material, Table S3 and presented in (A) and (B), respectively. ERG b/a-wave ratios for right and left eyes in each group are shown by open and shaded bars.
Genotype and influence on protein structure
All the missense mutations in the final data set have been reported recently (15) and are summarized in Table 1. These missense variants have been found to affect the following: (i) thiol group exchange, by insertion or removal of cysteine residues (variants C110Y, R141C and R200C); (ii) protein charge, by insertion, exchange or removal of charged residues without affecting thiol-containing residues (variants E72K, R102E, R102W, G109R, G140E, D168H, T185K, R191P, R197H and R213W); (iii) conformational stability of the polypeptide chain, by insertion or removal of proline residues (variants L69P, P192S, P203L and L216P) and (iv) hydrophobic core, by removal of the polar residue (variant G109W).
Structure-based analysis of 11 missense variants affecting the XLRS phenotype
| Mutation | CI score | Location and predicted effect of mutation |
|---|---|---|
| L69P | 0.36 | Buried hydrophobic leucine (ASA = 4 Å2) is replaced with a hydrophobic proline. This does not affect packing conditions in the hydrophobic core even if a Blosum70 score −3 is observed |
| R102E | 0.36 | The surface arginine residue located in the loop connecting β1 and β2 strands. The change from positively charged arginine to negatively charged glutamic acid is not severe (Bossum 70 score 0). This might potentially affect protein–protein interactions in the RS1 oligomer. Residue 102 is a ‘hot spot’ (three mutations) |
| G109R | 0.32 | Glycine is buried (ASA = 10 Å2). The mutation to arginine introduces a positive charge. Mild mutation with a negative Bossum70 score of −3. Residue 109 is the ‘hot spot’ (four mutations) |
| G109W | 0.45 | Buried glycine residue. This is a severe change which might affect the protein fold (Bossum70 score −3). Residue 109 is the ‘hot spot’ (four mutations) |
| C110Y | 0.48 | The cysteine 110 is buried. The mutation disrupts the intra-molecular disulphide bond C110-C142. The change decreases the stability of the hydrophobic core. This is a severe change which could affect the protein fold (Bossum70 score −3). Residue 110 is the ‘hot spot’ (two mutations) |
| G140E | 0.30 | Buried glycine residue (ASA = 0 Å2) is located in the loop between β1 and β2 strands. A mild substitution to negatively charged glutamic acid which does not cause a change in the domain stability |
| D168H | 0.26 | Buried residue is located in the loop connecting β4 and β5 strands. The ASA change is ΔASA = 9 Å2. This is a mild change with a Bossum70 score of −1 |
| T185K | 0.22 | The residue is located in the β6-strand and is exposed at the protein surface. This is a mild change which does not affect protein conformation |
| R191P | 0.33 | The missense change is mild |
| R197H | 0.36 | The residue is exposed (ASA = 53 Å2). This is a mild change which should not affect the protein fold. Residue 197 is the ‘hot spot’ (three mutations) |
| L216P | 0.38 | The buried hydrophobic leucine (ASA = 0 Å2) is replaced with hydrophobic proline. The missense change is mild |
| Mutation | CI score | Location and predicted effect of mutation |
|---|---|---|
| L69P | 0.36 | Buried hydrophobic leucine (ASA = 4 Å2) is replaced with a hydrophobic proline. This does not affect packing conditions in the hydrophobic core even if a Blosum70 score −3 is observed |
| R102E | 0.36 | The surface arginine residue located in the loop connecting β1 and β2 strands. The change from positively charged arginine to negatively charged glutamic acid is not severe (Bossum 70 score 0). This might potentially affect protein–protein interactions in the RS1 oligomer. Residue 102 is a ‘hot spot’ (three mutations) |
| G109R | 0.32 | Glycine is buried (ASA = 10 Å2). The mutation to arginine introduces a positive charge. Mild mutation with a negative Bossum70 score of −3. Residue 109 is the ‘hot spot’ (four mutations) |
| G109W | 0.45 | Buried glycine residue. This is a severe change which might affect the protein fold (Bossum70 score −3). Residue 109 is the ‘hot spot’ (four mutations) |
| C110Y | 0.48 | The cysteine 110 is buried. The mutation disrupts the intra-molecular disulphide bond C110-C142. The change decreases the stability of the hydrophobic core. This is a severe change which could affect the protein fold (Bossum70 score −3). Residue 110 is the ‘hot spot’ (two mutations) |
| G140E | 0.30 | Buried glycine residue (ASA = 0 Å2) is located in the loop between β1 and β2 strands. A mild substitution to negatively charged glutamic acid which does not cause a change in the domain stability |
| D168H | 0.26 | Buried residue is located in the loop connecting β4 and β5 strands. The ASA change is ΔASA = 9 Å2. This is a mild change with a Bossum70 score of −1 |
| T185K | 0.22 | The residue is located in the β6-strand and is exposed at the protein surface. This is a mild change which does not affect protein conformation |
| R191P | 0.33 | The missense change is mild |
| R197H | 0.36 | The residue is exposed (ASA = 53 Å2). This is a mild change which should not affect the protein fold. Residue 197 is the ‘hot spot’ (three mutations) |
| L216P | 0.38 | The buried hydrophobic leucine (ASA = 0 Å2) is replaced with hydrophobic proline. The missense change is mild |
An amino acid residue was considered to be located at the protein surface if the accessible surface area (ASA) was > 30 Å; otherwise, it was considered to be buried. Functional and structural effects for other seven missense variants: E72 K, R102W, R141C, P192S, P203L, R200C and R213W were described earlier (13) and are not included in the Table. Mutations with more severe changes are highlited. Hotspot mutation is defined as a number showing how many different mutations hit a same position in a protein structure.
Structure-based analysis of 11 missense variants affecting the XLRS phenotype
| Mutation | CI score | Location and predicted effect of mutation |
|---|---|---|
| L69P | 0.36 | Buried hydrophobic leucine (ASA = 4 Å2) is replaced with a hydrophobic proline. This does not affect packing conditions in the hydrophobic core even if a Blosum70 score −3 is observed |
| R102E | 0.36 | The surface arginine residue located in the loop connecting β1 and β2 strands. The change from positively charged arginine to negatively charged glutamic acid is not severe (Bossum 70 score 0). This might potentially affect protein–protein interactions in the RS1 oligomer. Residue 102 is a ‘hot spot’ (three mutations) |
| G109R | 0.32 | Glycine is buried (ASA = 10 Å2). The mutation to arginine introduces a positive charge. Mild mutation with a negative Bossum70 score of −3. Residue 109 is the ‘hot spot’ (four mutations) |
| G109W | 0.45 | Buried glycine residue. This is a severe change which might affect the protein fold (Bossum70 score −3). Residue 109 is the ‘hot spot’ (four mutations) |
| C110Y | 0.48 | The cysteine 110 is buried. The mutation disrupts the intra-molecular disulphide bond C110-C142. The change decreases the stability of the hydrophobic core. This is a severe change which could affect the protein fold (Bossum70 score −3). Residue 110 is the ‘hot spot’ (two mutations) |
| G140E | 0.30 | Buried glycine residue (ASA = 0 Å2) is located in the loop between β1 and β2 strands. A mild substitution to negatively charged glutamic acid which does not cause a change in the domain stability |
| D168H | 0.26 | Buried residue is located in the loop connecting β4 and β5 strands. The ASA change is ΔASA = 9 Å2. This is a mild change with a Bossum70 score of −1 |
| T185K | 0.22 | The residue is located in the β6-strand and is exposed at the protein surface. This is a mild change which does not affect protein conformation |
| R191P | 0.33 | The missense change is mild |
| R197H | 0.36 | The residue is exposed (ASA = 53 Å2). This is a mild change which should not affect the protein fold. Residue 197 is the ‘hot spot’ (three mutations) |
| L216P | 0.38 | The buried hydrophobic leucine (ASA = 0 Å2) is replaced with hydrophobic proline. The missense change is mild |
| Mutation | CI score | Location and predicted effect of mutation |
|---|---|---|
| L69P | 0.36 | Buried hydrophobic leucine (ASA = 4 Å2) is replaced with a hydrophobic proline. This does not affect packing conditions in the hydrophobic core even if a Blosum70 score −3 is observed |
| R102E | 0.36 | The surface arginine residue located in the loop connecting β1 and β2 strands. The change from positively charged arginine to negatively charged glutamic acid is not severe (Bossum 70 score 0). This might potentially affect protein–protein interactions in the RS1 oligomer. Residue 102 is a ‘hot spot’ (three mutations) |
| G109R | 0.32 | Glycine is buried (ASA = 10 Å2). The mutation to arginine introduces a positive charge. Mild mutation with a negative Bossum70 score of −3. Residue 109 is the ‘hot spot’ (four mutations) |
| G109W | 0.45 | Buried glycine residue. This is a severe change which might affect the protein fold (Bossum70 score −3). Residue 109 is the ‘hot spot’ (four mutations) |
| C110Y | 0.48 | The cysteine 110 is buried. The mutation disrupts the intra-molecular disulphide bond C110-C142. The change decreases the stability of the hydrophobic core. This is a severe change which could affect the protein fold (Bossum70 score −3). Residue 110 is the ‘hot spot’ (two mutations) |
| G140E | 0.30 | Buried glycine residue (ASA = 0 Å2) is located in the loop between β1 and β2 strands. A mild substitution to negatively charged glutamic acid which does not cause a change in the domain stability |
| D168H | 0.26 | Buried residue is located in the loop connecting β4 and β5 strands. The ASA change is ΔASA = 9 Å2. This is a mild change with a Bossum70 score of −1 |
| T185K | 0.22 | The residue is located in the β6-strand and is exposed at the protein surface. This is a mild change which does not affect protein conformation |
| R191P | 0.33 | The missense change is mild |
| R197H | 0.36 | The residue is exposed (ASA = 53 Å2). This is a mild change which should not affect the protein fold. Residue 197 is the ‘hot spot’ (three mutations) |
| L216P | 0.38 | The buried hydrophobic leucine (ASA = 0 Å2) is replaced with hydrophobic proline. The missense change is mild |
An amino acid residue was considered to be located at the protein surface if the accessible surface area (ASA) was > 30 Å; otherwise, it was considered to be buried. Functional and structural effects for other seven missense variants: E72 K, R102W, R141C, P192S, P203L, R200C and R213W were described earlier (13) and are not included in the Table. Mutations with more severe changes are highlited. Hotspot mutation is defined as a number showing how many different mutations hit a same position in a protein structure.
The majority of missense mutations had negative Blossom 70 scores (Table 1). This finding suggests that only a few missense mutations found in this study could potentially affect the stability of the RS structure, comparable with the effects of severe nonsense changes and protein truncations. The majority of missense mutations occurred in exons 4– 6 and some in exons 1–2. Most of the more severe mutations involved exon 1 and exons 3–6. RS1 is a secreted protein and the location of cysteine residues in the structure is thus important to the understanding of RS function (2,4,16–18). In the subjects with an altered cysteine residue, the alterations occurred in exons 5–6. The mature RS sequence consists of the Rs1 domain located in residues 25–64, and the discoidin domain occupying residues 65–224 (4). The mature RS1 structure contains 10 cysteines shared in an even proportion between Rs1 and discoidin domains. The positions of five cysteines are conserved within the protein family with discoidin domains (4,17,19). The other five cysteines are specific only for mature RS. To predict the location of these cysteines, a structure of mature RS was generated and refined using MD in water (13). The structure of the predicted discoidin domain of RS1 is consistent with an eight-strand distorted L-barrel motif (4,13,17,19). The expected functional roles for 11 new missense mutations are described in Table 1. Possible functions for the other seven mutations, which are not presented in Table 1, have previously been addressed (13,18).
Table 2 summarizes data from the 12 patients with alterations in RS1 splice sites, sequence duplications, exon deletions, insertions or deletions causing premature protein truncations. Five subjects had deletion of exon 1 and two subjects had splice site alterations disrupting coding at the intron–exon junction that might show no protein (null protein). Five subjects had DNA deletions or insertions, both likely to cause premature protein truncation.
Impact of missense changes
To understand how missense mutations affect the structure and stability of the protein molecule, residue accessibilities in the RS structure were analyzed in each mutant variant. Accessible surface area of the amino acid residue or surface accessibility is a measure of accessibility of the surface amino acid residues to the water environment surrounding protein. Accessible surface area could be used to determine changes at the protein surface caused by genetic mutations. Residues with accessibilities >30 Å2 were considered to be exposed at the protein surface; otherwise, residues were considered to be buried in the hydrophobic core (<30 Å2). The value of the CI score were in the range of 0.218 to 0.535 for mutations from T185K to R141C, respectively, with 14 mutations in class I and 4 in class II (Fig. 4A).
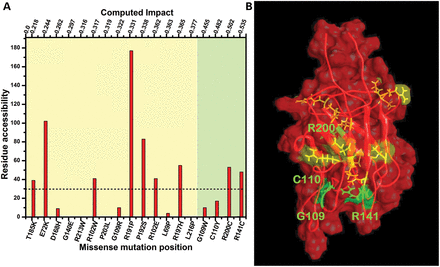
Localization of missense mutations in protein structure is shown in relation to severity. (A) Severity of structural change due to missense mutation was estimated by using the computed impact score (top scale); remutations considered as mild (0.22–0.43, light yellow) or as severe (0.435–0.535, light green). The amino acid residue was buried in the hydrophobic core if the residue accessibility was >30 Å2 (dashed horizontal line); otherwise the residue was considered as exposed at the surface (>30 Å2). (B) Structural model of RS1 shows the location of severe and mild mutations. Missense mutations causing a mild XLRS phenotype are shown in yellow. The severe mutations which are buried and/or related to the change from/to cysteine residue (green) are labeled. The molecular surface of protein is shown.
In the class II group, three missense variants were related to insertion or removal of cysteine residues and/or were buried in the hydrophobic core. A fourth mutation, G109W, was predicted to be a significant perturbation in a hydrophobic core, replacing a small glycine residue with a bulky hydrophobic tryptophan. Three severe mutations were located in the lower part of the molecule where structural elements, the so-called spikes, are localized (Fig. 4B). This is in keeping with previous data (13,16).
In contrast, the class I mutations were related to insertion, exchange or removal of charged residues without affecting cysteines and were divided into two equal subgroups of seven mutations. The first subgroup of mutations had accessibilities > 30Å2 and were classified as partially or fully exposed at the protein surface. Another subgroup of mutations show low or no accessibility and were predicted to be buried in the hydrophobic core. Some of these mutations are shown in Fig. 4B.
DISCUSSION
This study compares molecular modeling to the electrophysiological features in a cohort of 38 XLRS patients from Moorfields Eye Hospital (London, UK) having one of 18 missense variants. Patients were divided into groups based on mutation severity (mild; moderate; severe) and by age (<15, 15 to <30, 30 to <45, and 45+ years old). Molecular modeling predicted an association between mild and severe RS1 alterations and severity of phenotype that was consistent with ERG measures of inner retinal dysfunction in all but the oldest group of patients. The findings establish a genotype–phenotype correlation, which corroborates and extends the findings of an earlier study based on an independent cohort of XLRS patients (13).
The function of rod photoreceptors and the post-synaptic rod bipolar cell neurons is largely reflected in the amplitudes of the scotopic bright flash ERG a-wave (20,21) and b-wave (22–25), respectively, although there is a relatively small dark-adapted cone system contribution to this mixed rod and cone ERG (26). In most cases of XLRS, the dark-adapted full-field ERG to a bright white flash shows an abnormal ‘electronegative waveform’, where the b-wave is of lower amplitude than the negative polarity a-wave (27); in normal subjects, the b-wave amplitude is greater than the a-wave (28). Although substantial ERG variability is observed (27,29), the amplitude of the a-wave is reduced in up to 30% of XLRS patients (27). There is also evidence from the RS1-knockout mouse that the disease process affects rod photoreceptors with a loss of outer nuclear layer cells with age (14,30,31). In the current study, the ERG b/a-wave ratio provided a measure of inner retinal function largely independent of possible additional abnormalities resulting in significant a-wave reduction. Age-related differences in dark-adapted ERG parameters were consistent with those reported previously in the RS1-knockout mouse.
In previous work (13), CI values were chosen to separate mutations into severe and less severe groups, which correctly explained the secretion of RS1 in 85% of 27 mutations. However, information on protein secretion in cell culture could not be simply transferred to clinical cases. In the current study, the classification was refined by introducing a moderate (intermediate) CI group associated with a relatively wide range of ERG phenotypes, possibly related to the role of proline or arginine residues (Supplementary Material, Table S4). The remaining ERG data could be divided into one of two quantitatively distinct ERG phenotypes. The majority of class I (mild) mutations showed no changes involving cysteine residues, were evenly distributed between the RS1 surface and the hydrophobic core, and were associated with mutations in the water inaccessible residues when changes of polar-to-polar or the hydrophobic-to-hydrophobic have occurred (Fig. 5). Class II (severe) mutations caused severe perturbations due to the removal or insertion of cysteine residues or due to changes in the hydrophobic core.
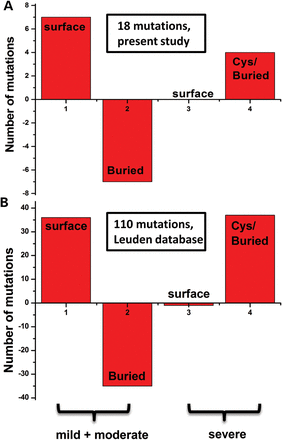
A link between phenotypic severity of missense variant and the mutation location in the RS1 three-dimensional structure is shown. The severity of structural change due to missense mutation was estimated using the computed impact score. Residues were divided in two groups by computed severity value, CI, and considered as mild (CI ≤ 0.4) or as severe (CI > 0.4). (A) A group of 18 missense variants collected in the Moorfields Eye Hospital. (B) Computed severity of missense mutations was calculated by the method similar to that described by Sergeev et al. (2010) for 110 missense variants causing X-linked retinoschisis, which were acquired from the Leiden Open Variation Database (http://grenada.lumc.nl/LOVD2/eye/home.php) and the Retina International Newsletter Mutation Database (http://www.retina-international.com/sci-news/mutation.htm). Positive and negative numbers at the Y-axis demonstrate consistency or inconsistency, respectively, with results obtained according to predictions from Sergeev et al. Numbers at the X-axis show results for mild, moderate (1, 2) and severe (3, 4) mutations, respectively.
Genotype–phenotype relationships
Previous studies have investigated genotype–phenotype relationships in XLRS but missense, splice site, frameshift, insertion and deletion mutations were reported to result in similar phenotypes (32–34). No correlation was found between disease severity characterized by visual acuity (33,35–39) and mutation type, either for small patient numbers or in a larger study of 86 XLRS patients in whom causative RS1 mutations were identified (40). More recently, detailed ERG data from the current cohort demonstrated that nonsense, splice site or frame-shifting mutations in RS1 consistently result in severe ERG changes characterized by a markedly negative bright flash ERG, delayed flicker response and abnormal pattern ERG (15). Missense mutations resulted in a wider range of ERG abnormalities and included those with mild and also severe ERG phenotypes indistinguishable from the non-missense group. The current study shows that these phenotypic extremes may be predicted according to the CI of different missense variants.
Protein stability and missense changes
An interesting implication, when using computed severity scores, is that mutations in the hydrophobic core and/or affecting cysteines are more severe than mutations on the protein surface. Indeed, in the endoplasmic reticulum (ER) proteins fold into their native conformations and undergo different post-translational modifications and the formation of disulphide bonds. In the ER, the formation of a disulphide bond is catalyzed by protein disulphide isomerase (PDI), a cellular chaperone with foldase function to maintain a native protein fold (41,42). PDI catalyzes oxidation and reshuffling (isomerization) of disulphides in the substrate proteins motif (43).
Because the ‘energy surface’ or the ‘landscape’ is encoded by the amino-acid sequence (44), the protein folds using several competing pathways into intermediate non-native structures with decreasing free energies until it achieves a conformation with the lowest energy to form a protein with native interactions (45). Misfolded or intermediate non-native proteins never achieve this lowest energy minimum. Pathogenic missense mutations might inhibit the pathway to the lowest energy conformation and cause the protein to stay in an intermediate or misfolded conformation with a higher free energy. Misfolded proteins could be a subject of chaperone-mediated autophagy (46) and/or they might be delivered to the cytosol for degradation in the ubiquitin–proteasome pathway (47). This is confirmed by numerous observations on RSs modified by severe mutations and expressed in cell cultures that show the modified RS1 bands to be absent on SDS–PAGE or native gels and associated with a loss of protein due to misfolding and rapid degradation (4,16–18), similar to that of null mutations (48).
RS secretion
RS is secreted as a homo-oligomeric complex in vivo in photoreceptors and bipolar cells; proper secretion of a functional RS1 could be interrupted by protein misfolding caused by pathogenic mutation (4,16,17,18).
In previous work using a CI score, all missense mutations were divided into two groups by their ability to secrete a functional octamer, and the observed secretion of octamers and predictions using a CI score were an exact match for 85% of all mutations analyzed (13). Thus, a relative success in correct predictions suggests that the CI score provided a useful technique to rank missense mutations by the degree of structural severity and, particularly, classify missense variants by their ability to form homo-octamers in vivo. Indeed, within the 18 missense mutations in the currently analyzed data set of missense variants, E72K, G140E, D168H and T185K demonstrate mild changes (class I) and are predicted to show a mutant variant secretion (CI < 0.3); the remaining Class II severe missense variants are expected to show no secretion.
‘Hot spot’ mutations and putative protein-binding surfaces
Currently, ∼110 missense mutations are implicated in XLRS. This includes several mutations that target the same position in the protein sequence. These ‘hotspot’ sites are located in several areas of the protein sequence as shown in Fig. 6, Supplementary Material, Table S5 and Supplementary Data. Eleven of the 18 mutations from the Moorfields' data set could be considered as the hotspot sites. The severe mutations, as predicted using the CI index >0.4, are located in ‘hot’ sites with two or more mutations in the same position: G109W (0.45), C110Y (0.48), R141C (0.54) and R200C (0.50). Hotspot mutations are concentrated in certain ‘hottest’ areas at the protein surface, which are associated with ligand-binding activity (spikes 1–3, A) and protein–protein surface interactions in oligomeric complex (B), and determine native conformation of the RS1 protein fold (termini residues, C) (Fig. 6 insets). Some of these areas might be potentially involved in protein–protein interactions as was shown for the predicted structure of the RS1 dimer (Fig. 7). This suggests that hotspot surface areas potentially are important for proper function of RS1 and, in part, could be involved in the formation of the protein–protein interface to maintain the RS1 oligomeric state (16,17).
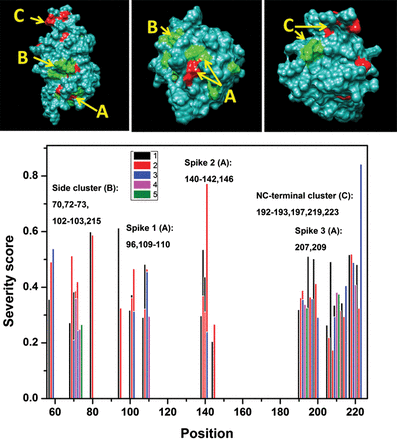
Predicted severities of 110 pathogenic missense changes at positions of the protein sequence show the areas affected most by hotspot mutations. These areas are shown at the top three insets by red (severe) and green (mild or moderate). Surface areas of spikes, side cluster and NC-terminal cluster are labeled by (A), (B) and (C), respectively. Bottom panel: Up to five individual predicted severities (CI values) are shown for each hotspot by five different colors. In each sequence position the number of hotspot mutations is shown in black (110 missense changes) and red (present study) (Supplementary Material, Fig. S2).
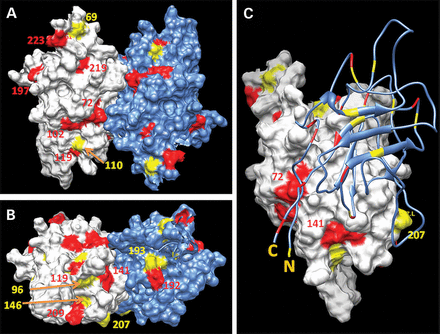
A significant fraction of hotspot mutations is localized to the protein surface. Two different views of the accessible surface area of the RS1 dimer, front and bottom, where the majority of hotspot mutations are concentrated, are shown in (A) and (B), respectively. Related by 2-fold symmetry molecules of the dimer are shown in white (molecule A) and blue (molecule B). (C) The molecule B is shown by a blue ribbon to demonstrate a contact surface between subunits within a dimer interface. Surface areas with two or three to five spots at the same sequence position of the structure are shown in yellow and red, respectively. Sequence positions of hotspot mutations at the protein surface are shown with the corresponding colors.
In summary, the data suggest that missense changes in RS1 can be divided into mild and severe classes that have a markedly different impact on RS structure and function. The study shows that severe phenotypes are associated with maximum structural perturbations that can relate to dramatic changes in the protein hydrophobic core or to the deletion or insertion of cysteine residues that affect in general the stability of protein folding or failure to be secreted. The grouping helps predict the severity of ERG abnormalities relating to global inner retinal dysfunction and may influence patient management and the selection of candidates for possible future therapeutic interventions. This is the second study of XLRS, using a completely independent patient cohort, to indicate a genotype–phenotype correlation.
In general, the ability to establish genotype-to-phenotype relationships could be used to assess disease risk using atomic models of proteins for a broad spectrum of inherited eye disorders. The prediction of a functional phenotype (b/a-ratios) from the patient genotype potentially could be useful in clinical trials or other clinical studies to identify patient groups with severe and less severe phenotypes and might provide some rationale to choose medical treatment for each group. Indeed, the small molecule drug treatment might be more appropriate for patients with mild missense changes. In contrast, gene therapy could be suggested as a treatment to compensate the effect of the ‘null’ protein caused by the severe missense change. Finally, a combination of information about the structural perturbations of protein caused by pathogenic mutations at the atomic level, computational drug design and phenotype classification might improve a search for new therapeutics in inherited eye disease.
MATERIALS AND METHODS
The basis of this study was a group of 57 patients with XLRS who were examined and genotyped at Moorfields Eye Hospital, London, UK (15). One of the important aspects of this cohort is that all electrophysiology was performed at a single site using a standardized protocol. International-standard full-field ERGS (30) were available for analysis in 50 of these patients, including 38 cases with 18 missense variants and 12 cases with non-missense or null mutations. For the purposes of the current study, disease severity was estimated by measuring the scotopic bright flash (DA 11.0) ERG and single flash cone (LA 3.0) ERG a- and b-wave parameters. The ratio of ERG b/a-wave amplitudes was used as a measure of generalized inner retinal dysfunction (49).
RS1 mutation analysis
The impact on RS protein structure by particular missense mutation was assessed computationally (13). Patients were classified into one of three groups based on the CI score: mild (<0.32; ‘Class I’), moderate (0.32 to <0.4; ‘Intermediate’) and severe (>0.4; ‘Class II’). Patients were grouped into one of four age groups: <15, 15 to <30, 30 to <45, 45+ years. Genetic mutations were classified as severe if associated with a significant change or elimination of RS1 expression, as in the 12 cases identified with non-missense mutations related to alterations in splice sites, sequence duplications, exon deletions, insertions or deletions (Table 2). Computed impact (CI) scores for missense variants were compared with the severity of inner retinal dysfunction as measured by the full-field ERG parameters.
Structure of RS and computed severity of missense changes
The structure of mature human RS was modeled by homology as described previously (13). Briefly, the X-ray structure of the C2 domain of FaV was chosen as the structural template (Brookhaven protein database, PDB code: 1sdd, chain B4) (50). The RS1 protein structure was built by the automatic segment matching method (51) in the program Gene Mine Look, version 3.5.2 (52), followed by 500 cycles of energy minimization. Hydrogens were added to the RS1 structure and the structure was regularized by an energy minimization procedure in the presence of water using the Impact module of the Maestro program package (version 8.0.308, Schrodinger Inc., New York). MD trajectories were calculated in a periodic rectangular box (100 Å × 50 Å × 50 Å) of 6897 explicit SPC water molecules. The structure of the RS1 dimer was modeled using the dimeric structure of the membrane-binding discoidin-like C2 domain of human coagulation factor V (5).
The structure of the MD-equilibrated RS1 was used to generate in silico the structural models of proteins modified by missense variants. The program Look, version 3.5.2, was employed to generate and refine the conformation of missense variants (53). Predicted structures of each mutant protein were regularized by an energy minimization procedure as described above for the native RS1 in the presence of water for the final step. Finally, atomic structures were generated for L69P, R102E, G109R, G109W, C110Y, G140E, D168H, T185K, R191P, R197H and L216P missense variants associated with XLRS to evaluate the computed severities caused by missense changes.
Mutated residue accessibility was calculated by the surface accessibility algorithm (54,55). The amino acid residue was considered as buried in the hydrophobic core if the residue accessibility was less than 30 Å2 (dashed horizontal line); otherwise the residue was considered as exposed at the surface (>30 Å2).
The rapid evaluation of computed severities due to missense mutations was accessed using the FoldX force field (56) and the Grantham difference (57) for each amino acid pair to characterize physiochemical differences between wild-type and missense variant amino acid residues, as suggested previously (13). The CI score was calculated for each mutation and compared with the severity of inner retinal dysfunction measured as the ERG b/a-wave amplitude ratio.
Statistical analysis
Statistical analysis was performed using analysis software (ProcGENMOD, version 9.2; SAS Institute, Cary, NC, USA) similar to that described previously (58). Briefly, analyses included both eyes of every subject, adjusted for intrapersonal correlation (Generalized Estimating Equation Model) without making any specific assumptions about the structure of the correlation. The severity of missense change was evaluated using the molecular grading scale, which is characterized by the CI index (13). Continuous linear regression and correlation coefficients were computed across all ages or across the severity of mutational change of a given group. Analysis was performed according to the four age groups and three classes of missense change. In a population of healthy eyes, ERG b-wave amplitudes are not normally distributed (59,60) and often are transformed to meet the assumptions of most statistical analyses. A log transform was performed on the a-wave and b-wave amplitudes prior to statistical analysis.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by National Institutes of Health (Z01-EY000476-01 to Y.V.S., and Z01-DC000065-08 to P.A.S.) and Foundation Fighting Blindness (USA), and was partly funded by the National Institute for Health Research UK( NIHR)Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology.
ACKNOWLEDGEMENTS
We are thankful for the use of the commercial molecular modeling software on the network through the Center of Molecular Modeling, CIT/NIH.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}